Intro_FindIT2:peakGeneCor module
在上一篇文章中,我介绍了如何用mmAnno模块中的各个函数建立peak与gene之间的link关系。但实际上,不管用什么样的注释策略,都基于的是geomic location上的关系。除了genomic location之外,我们还可以利用peak开放变化与gene表达变化的一致关系来建立link关系,其本质就是计算两者的相关性,如果peak与对应基因的变化一致性越高,那么越有可能说明peak是调控这个基因的。这个策略往往被用来挖掘调控感兴趣基因的远端元件,在样本量足够大的情况下,往往可以达到Hi-C的准确度12。正是基于此,我写了第二个模块:peakGeneCor module,来方便具有多样本ATAC-seq数据的用户来进一步地挖掘元件与基因的关系。peakGeneCor module有两个函数:peakGeneCor和enhancerPromoterCor,前者是用来建立peak与基因的相关性,而后者是用来建立远端enhancer与近端promoter的相关性。具体的细节我会在后面描述
Pre-preparation
这次的测试数据仍旧来自于FindIT2包本身,包含一个ATAC的mergePeak文件、一组ATAC count矩阵和一组RNA-seq count矩阵。首先我们需要加载对应的包以及数据。
# 加载包
library(FindIT2)
library(TxDb.Athaliana.BioMart.plantsmart28)
library(ggplot2)
Txdb <- TxDb.Athaliana.BioMart.plantsmart28
seqlevels(Txdb) <- paste0("Chr", c(1:5, "M", "C"))# 读取ATAC merge peak文件
# 为了加快测试,我这里只用了前100个peak
peak_path <- system.file("extdata", "ATAC.bed.gz", package = "FindIT2")
peak_GR <- loadPeakFile(peak_path)[1:100]
# 再次注意,请一定要有feature_id这一列
peak_GR
## GRanges object with 100 ranges and 1 metadata column:
## seqnames ranges strand | feature_id
## <Rle> <IRanges> <Rle> | <character>
## [1] Chr5 27-311 * | WFX_peak_19629
## [2] Chr5 501-2153 * | WFX_peak_19630
## [3] Chr5 2611-2965 * | WFX_peak_19631
## [4] Chr5 5055-5611 * | WFX_peak_19632
## [5] Chr5 8093-10183 * | WFX_peak_19633
## ... ... ... ... . ...
## [96] Chr5 346146-346877 * | WFX_peak_19724
## [97] Chr5 348523-348794 * | WFX_peak_19725
## [98] Chr5 349128-349354 * | WFX_peak_19726
## [99] Chr5 357097-358081 * | WFX_peak_19727
## [100] Chr5 358865-359472 * | WFX_peak_19728
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
# 加载矩阵数据
data("RNA_normCount")
data("ATAC_normCount")
# ATAC和RNA-seq都是7个时间点的样本,每个时间点里面有2-3个样本
head(RNA_normCount)
## E5_0h_R1 E5_0h_R2 E5_4h_R1 E5_4h_R2 E5_4h_R3 E5_8h_R1
## AT5G00730 0.000000 0.000 0.000 0.000 0.000 0.0000000
## AT5G01010 7248.089928 8403.591 3377.432 2813.139 2283.258 3964.8175573
## AT5G01015 1.892452 0.000 0.000 0.000 0.000 0.8457375
## AT5G01017 0.000000 0.000 0.000 0.000 0.000 0.0000000
## AT5G01020 3169.856561 2460.302 1569.942 1519.461 1500.550 1053.7889668
## AT5G01030 1566.949990 2124.571 1888.195 1592.688 1196.404 1050.4060167
## E5_8h_R2 E5_16h_R1 E5_16h_R2 E5_24h_R1 E5_24h_R2 E5_48h_R1
## AT5G00730 0.0000 0.000 0.0000 0.0000000 0.0000 0.000000
## AT5G01010 3416.2812 2939.066 2749.5849 2331.0450812 2338.7608 2257.904991
## AT5G01015 0.0000 0.000 0.0000 0.5954138 0.0000 4.532808
## AT5G01017 0.0000 0.000 0.0000 0.0000000 0.0000 0.000000
## AT5G01020 1612.5888 1740.990 1672.8432 1768.9744409 1891.6211 2363.292777
## AT5G01030 979.1008 1022.450 817.6256 853.8234090 860.4622 646.491743
## E5_48h_R2 E5_72h_R1 E5_72h_R2 E5_72h_R3
## AT5G00730 0.000000 0.000000 0.000000 0.000
## AT5G01010 2451.523394 2076.693944 1889.352383 1929.887
## AT5G01015 2.072294 4.663235 3.024577 0.000
## AT5G01017 0.000000 0.000000 0.000000 0.000
## AT5G01020 2189.032864 2647.163013 2699.938998 2705.583
## AT5G01030 714.941311 1122.285200 1125.142615 977.176
head(ATAC_normCount)
## E5_0h_R1 E5_0h_R2 E5_4h_R1 E5_4h_R2 E5_8h_R1 E5_8h_R2
## WFX_peak_19629 96.44832 87.34256 145.75031 95.57316 137.46488 101.17315
## WFX_peak_19630 377.19278 339.05895 440.10878 351.79057 443.53902 478.44498
## WFX_peak_19631 54.67452 41.85164 37.15204 48.80332 48.32750 49.16826
## WFX_peak_19632 168.93814 163.76729 162.89740 160.64425 204.04943 142.77706
## WFX_peak_19633 653.63700 665.98700 625.86897 679.17948 548.78557 638.24183
## WFX_peak_19634 101.36288 60.65455 37.15204 65.07109 50.47539 57.67815
## E5_16h_R1 E5_16h_R2 E5_24h_R1 E5_24h_R2 E5_48h_R1 E5_48h_R2
## WFX_peak_19629 101.67500 150.53297 141.34424 107.42046 93.89768 107.17680
## WFX_peak_19630 416.13378 504.86443 555.53209 499.18920 359.02054 433.26790
## WFX_peak_19631 73.37371 97.26746 78.05577 67.78403 41.97779 63.85001
## WFX_peak_19632 191.81985 222.32562 198.30386 198.75658 177.85325 158.48484
## WFX_peak_19633 769.37581 788.56119 736.25582 711.73236 501.52408 556.40719
## WFX_peak_19634 80.71109 78.74032 84.38462 80.99618 96.10704 49.02768
## E5_48h_R3 E5_72h_R1 E5_72h_R2 E5_72h_R3
## WFX_peak_19629 102.17173 81.22392 77.57811 79.00373
## WFX_peak_19630 374.90435 339.76767 363.39219 427.93688
## WFX_peak_19631 84.86846 53.76795 44.91364 62.91038
## WFX_peak_19632 175.50467 137.27987 154.13545 178.48991
## WFX_peak_19633 530.63385 498.78352 576.73198 606.42679
## WFX_peak_19634 60.14949 52.62395 41.85135 105.33831由于我们后面会用到几个Shiny函数,所以需要额外再加载一个叫InteractiveFindIT2的包。这个包里面有两个辅助的Shiny函数。
原来这两个Shiny函数是在FindIT2包里面的,不过reviewer要求我将这两个函数剥离出来保证整个包的功能专一性,于是我就重新写了InteractiveFindIT2。由于我的Shiny水平很捉急,所以这个包非常的“丑陋”,但还是可以凑合着用的。
library(InteractiveFindIT2)peakGeneCor
peakGeneCor可以接受mm_nearestGene和mm_geneSacn产生的注释结果作为输入。其本质上就是根据你注释得到的peak-gene link,去寻找相应的开放和表达变化,然后用R语言的内置函数cor.test去计算pearson相关系数及对应的显著性。
首先我们用mm_geneScan去注释我们这100个peak。因为我们这里想要看下是否有些远端元件跟基因表达是呈现相关性的,所以我们可以把scan范围设的大一点,这样我们可以尽可能地包含更多的distal peak-gene link。当然,如果你只是单纯地想要看下你的peak跟基因的相关性,也可以用mm_nearestGene的结果作为输入。
mmAnno <- mm_geneScan(peak_GR = peak_GR,
Txdb = Txdb,
upstream = 2e4,
downstream = 2e4)
## >> checking seqlevels match... 2021-08-16 19:20:44
## >> your peak_GR seqlevel:Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## ------------
## annotatePeak using geneScan mode begins
## >> preparing gene features information and scan region... 2021-08-16 19:20:44
## >> some scan range may cross Chr bound, trimming... 2021-08-16 19:20:45
## >> finding overlap peak in gene scan region... 2021-08-16 19:20:45
## >> dealing with left peak not your gene scan region... 2021-08-16 19:20:45
## >> merging two set peaks... 2021-08-16 19:20:45
## >> calculating distance and dealing with gene strand... 2021-08-16 19:20:45
## >> merging all info together ... 2021-08-16 19:20:45
## >> done 2021-08-16 19:20:45# 可以看到,产生了非常多的peak-gene link,是peak数目的10倍之多
mmAnno
## GRanges object with 1204 ranges and 3 metadata columns:
## seqnames ranges strand | feature_id gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] Chr5 27-311 * | WFX_peak_19629 AT5G01010
## [2] Chr5 501-2153 * | WFX_peak_19630 AT5G01010
## [3] Chr5 2611-2965 * | WFX_peak_19631 AT5G01010
## [4] Chr5 5055-5611 * | WFX_peak_19632 AT5G01010
## [5] Chr5 8093-10183 * | WFX_peak_19633 AT5G01010
## ... ... ... ... . ... ...
## [1200] Chr5 358865-359472 * | WFX_peak_19728 AT5G01960
## [1201] Chr5 357097-358081 * | WFX_peak_19727 AT5G01970
## [1202] Chr5 358865-359472 * | WFX_peak_19728 AT5G01970
## [1203] Chr5 357097-358081 * | WFX_peak_19727 AT5G01980
## [1204] Chr5 358865-359472 * | WFX_peak_19728 AT5G01980
## distanceToTSS
## <numeric>
## [1] 4749
## [2] 2907
## [3] 2095
## [4] 0
## [5] -3031
## ... ...
## [1200] -10927
## [1201] 16872
## [1202] 15481
## [1203] -17133
## [1204] -15742
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths在得到了peak-gene link之后,我们还需要再整理下我们的ATAC和RNA-Seq矩阵。因为计算相关性的时候需要对应的时间点,比如说ATAC的0h对应RNA的0h,4h对应4h。但我们这里拿到的是每个时间点内部的生物学重复数据,所以我们还要取下平均值,从而来得到对应的7个时间点的数据。
针对上面这种情况,FindIT2提供了一个辅助函数integrate_replicates来帮助用户更方便地计算平均值。
除了求平均之外,
integrate_replicates还有很多有用的功能,后面会一一介绍。
# 首先我们需要建立一个coldata数据框来表明你各个重复对应的处理/时间点
# 注意integrate_replicates函数只接受一列的数据框,且列名叫type,请不要再多加其他的信息。
ATAC_colData <- data.frame(
row.names = colnames(ATAC_normCount),
type = gsub("_R[0-9]", "", colnames(ATAC_normCount))
)
head(ATAC_colData)
## type
## E5_0h_R1 E5_0h
## E5_0h_R2 E5_0h
## E5_4h_R1 E5_4h
## E5_4h_R2 E5_4h
## E5_8h_R1 E5_8h
## E5_8h_R2 E5_8h
ATAC_normCount_merge <- integrate_replicates(ATAC_normCount, ATAC_colData)
head(ATAC_normCount_merge)
## E5_0h E5_4h E5_8h E5_16h E5_24h E5_48h
## WFX_peak_19629 91.89544 120.66173 119.31901 126.10399 124.3824 101.08207
## WFX_peak_19630 358.12587 395.94967 460.99200 460.49911 527.3606 389.06426
## WFX_peak_19631 48.26308 42.97768 48.74788 85.32059 72.9199 63.56542
## WFX_peak_19632 166.35271 161.77083 173.41325 207.07274 198.5302 170.61425
## WFX_peak_19633 659.81200 652.52423 593.51370 778.96850 723.9941 529.52171
## WFX_peak_19634 81.00872 51.11156 54.07677 79.72571 82.6904 68.42807
## E5_72h
## WFX_peak_19629 79.26859
## WFX_peak_19630 377.03225
## WFX_peak_19631 53.86399
## WFX_peak_19632 156.63508
## WFX_peak_19633 560.64743
## WFX_peak_19634 66.60454RNA_colData <- data.frame(
row.names = colnames(RNA_normCount),
type = gsub("_R[0-9]", "", colnames(RNA_normCount))
)
head(RNA_colData)
## type
## E5_0h_R1 E5_0h
## E5_0h_R2 E5_0h
## E5_4h_R1 E5_4h
## E5_4h_R2 E5_4h
## E5_4h_R3 E5_4h
## E5_8h_R1 E5_8h
RNA_normCount_merge <- integrate_replicates(RNA_normCount, RNA_colData)
head(RNA_normCount_merge)
## E5_0h E5_4h E5_8h E5_16h E5_24h E5_48h
## AT5G00730 0.0000000 0.000 0.0000000 0.0000 0.0000000 0.000000
## AT5G01010 7825.8406557 2824.609 3690.5493982 2844.3252 2334.9029648 2354.714192
## AT5G01015 0.9462258 0.000 0.4228688 0.0000 0.2977069 3.302551
## AT5G01017 0.0000000 0.000 0.0000000 0.0000 0.0000000 0.000000
## AT5G01020 2815.0790782 1529.984 1333.1889011 1706.9164 1830.2977816 2276.162820
## AT5G01030 1845.7605682 1559.096 1014.7534263 920.0376 857.1427856 680.716527
## E5_72h
## AT5G00730 0.000000
## AT5G01010 1965.310975
## AT5G01015 2.562604
## AT5G01017 0.000000
## AT5G01020 2684.228337
## AT5G01030 1074.867934在得到了平均值矩阵之后,我们就可以计算每个link里面peak和对应基因的相关性了。需要注意的是,除非你的mmAnno的行数非常多(比如说上百万),不然请不要把parallel设置为TRUE。因为在执行并行化的时候,资源的调配也是需要时间的,对于少量的mmAnno执行并行化来计算相关性反而会拖累你的速度。
mmAnnoCor_peakGene <- peakGeneCor(mmAnno = mmAnno,
peakScoreMt = ATAC_normCount_merge,
geneScoreMt = RNA_normCount_merge,
parallel = FALSE)
## Good, your two matrix colnames matchs
## Warning: some gene_id or feature_id in your mmAnno is not in your geneScoreMt or peakScore Mt, final cor and pvalue of these gene_id or feature_id pair will be NA
## >> calculating cor and pvalue, which may be time consuming... 2021-08-16 19:20:46
## >> merging all info together... 2021-08-16 19:20:46
## >> done 2021-08-16 19:20:46可以看到,产生的相关性结果依然是一个GRanges对象。peakGeneCor函数只不过是在你输入的注释后面又额外添加了相关性系数、显著性等操作。这样用户就可以方便地查看peak跟gene的变化关系了。
mmAnnoCor_peakGene
## GRanges object with 1204 ranges and 7 metadata columns:
## seqnames ranges strand | feature_id gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] Chr5 27-311 * | WFX_peak_19629 AT5G01010
## [2] Chr5 27-311 * | WFX_peak_19629 AT5G01015
## [3] Chr5 27-311 * | WFX_peak_19629 AT5G01020
## [4] Chr5 27-311 * | WFX_peak_19629 AT5G01030
## [5] Chr5 27-311 * | WFX_peak_19629 AT5G01040
## ... ... ... ... . ... ...
## [1200] Chr5 358865-359472 * | WFX_peak_19728 AT5G01940
## [1201] Chr5 358865-359472 * | WFX_peak_19728 AT5G01950
## [1202] Chr5 358865-359472 * | WFX_peak_19728 AT5G01960
## [1203] Chr5 358865-359472 * | WFX_peak_19728 AT5G01970
## [1204] Chr5 358865-359472 * | WFX_peak_19728 AT5G01980
## distanceToTSS cor pvalue padj qvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 4749 -0.244462 0.59727694 1 0.739428
## [2] 5579 -0.750863 0.05175846 1 0.386725
## [3] 8132 -0.893576 0.00669623 1 0.226781
## [4] -9557 -0.241763 0.60145919 1 0.739428
## [5] 15861 0.222311 0.63184956 1 0.744406
## ... ... ... ... ... ...
## [1200] 5296 -0.182663 0.695048 1 0.762868
## [1201] 10713 0.486188 0.268609 1 0.599203
## [1202] -10927 0.184569 0.691975 1 0.762868
## [1203] 15481 -0.362072 0.424812 1 0.663994
## [1204] -15742 -0.517337 0.234387 1 0.576202
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths对于产生的mmAnnoCor_peakGene,FindIT2还提供了一个画图函数来帮助用户展现感兴趣的基因。这里我随便挑了一个AT5G01010基因,从下图可以看到,该基因周围大部分peak跟其相关性都不是很高,只有一个较远的peak:WFX_peak_19367跟其相关性比较高。
plot_peakGeneCor(mmAnnoCor = mmAnnoCor_peakGene,
select_gene = "AT5G01010") -> p
## Joining, by = "feature_id"
## Joining, by = "feature_id"
p
## `geom_smooth()` using formula 'y ~ x'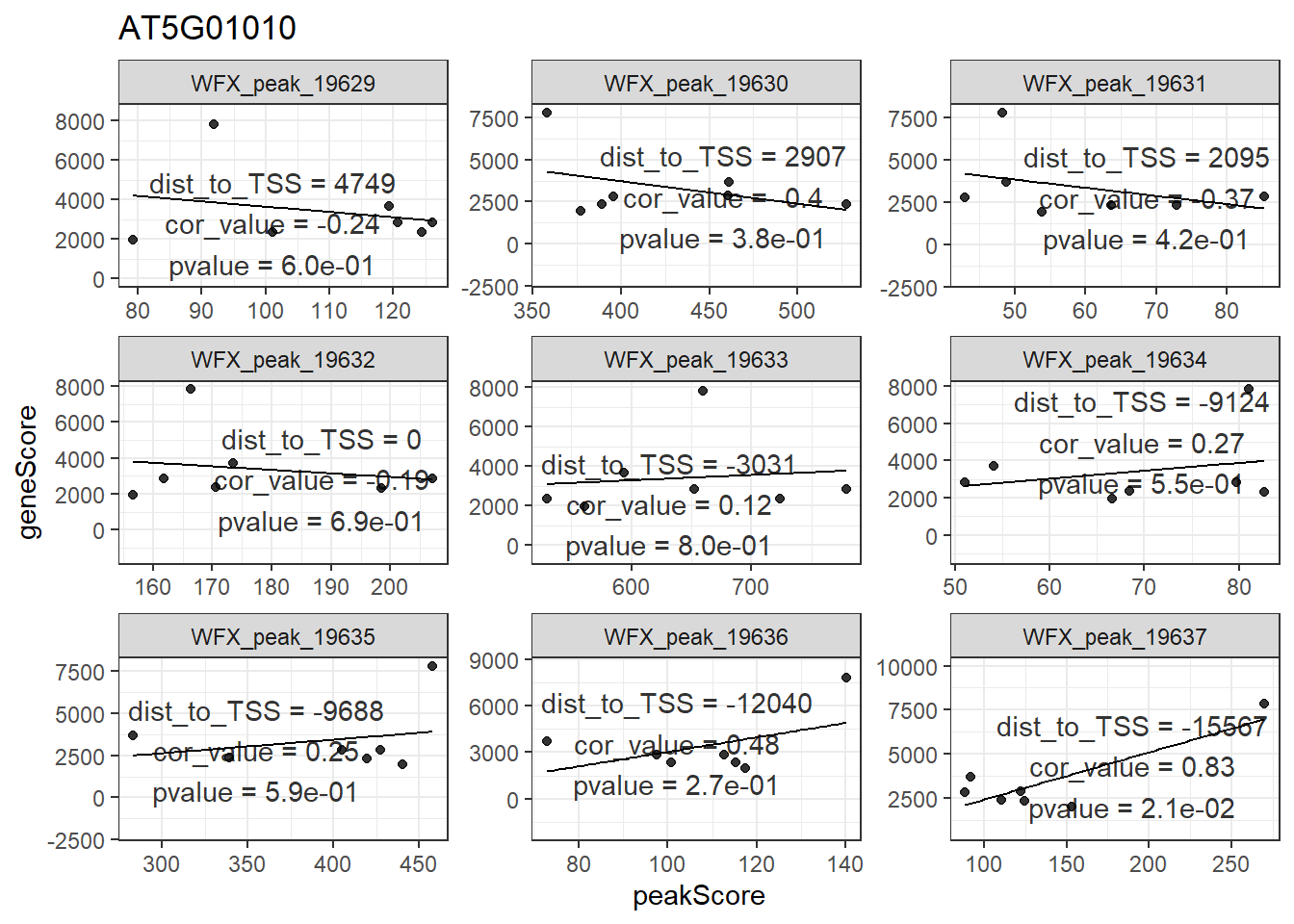
因为这产生的是ggplot2的对象,如果你想要展示颜色,也可以ggplot2的语法加上去。
p +
geom_point(aes(color = time_point))
## `geom_smooth()` using formula 'y ~ x'
除了自己选择基因之外,我们还可以用InteractiveFindIT2包里面的shinyParse_peakGeneCor函数来交互式地选择基因进行探究。你可以在Gene栏选择基因,在Cor_filter和pvalue_filter栏设置筛选条件,然后点击plot,即可出现图片。
shinyParse_peakGeneCor(mmAnnoCor = mmAnnoCor_peakGene)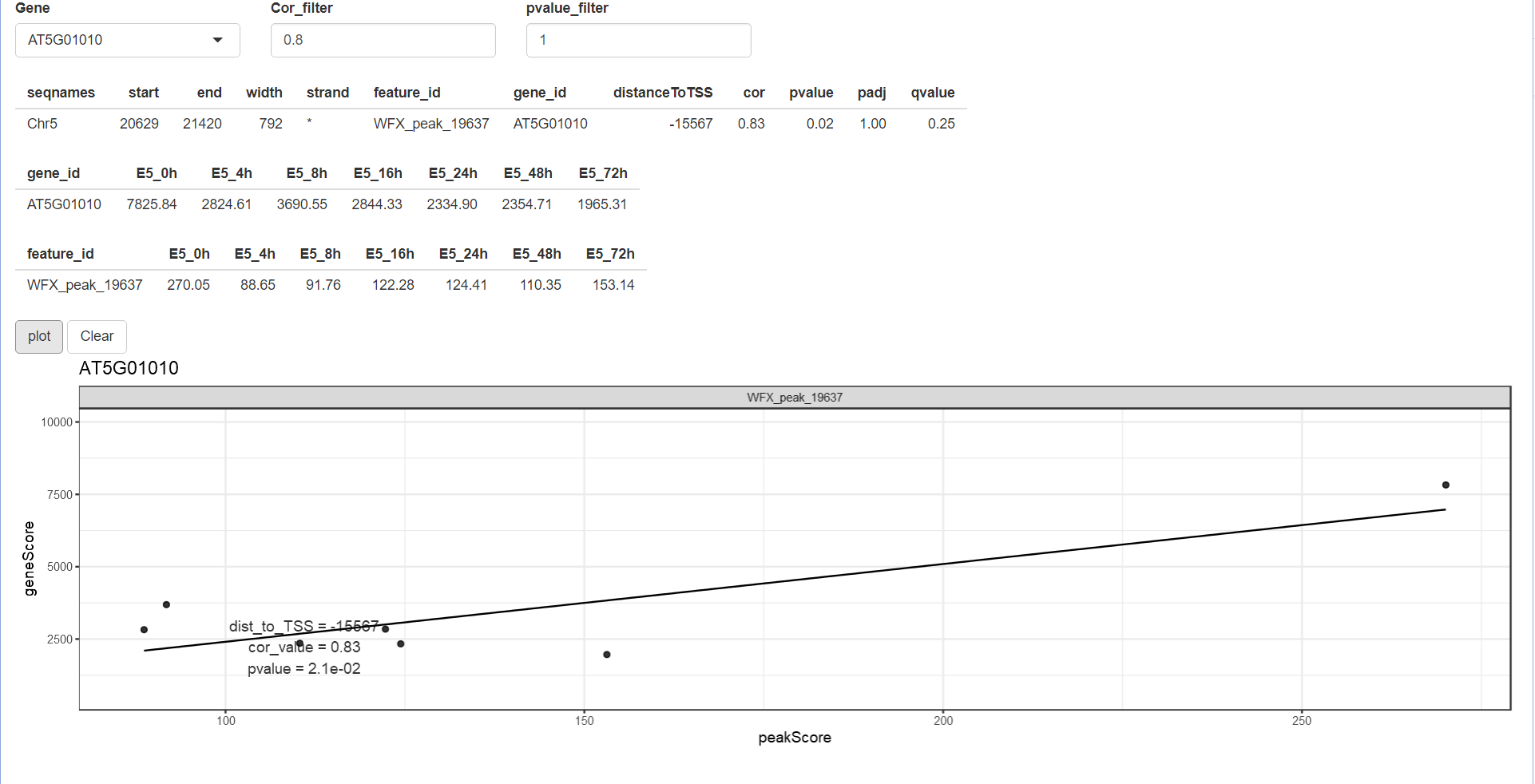
enhancerPromoterCor
除了前面的peakGeneCor函数,FindIT2还提供了另一个函数enhancerPromterCor。这个函数的由来有这么几个原因:
首先是样本量的原因。前面提到
peakGeneCor计算的是peak与基因的相关性,本质上是基于两者变化量的一致性,用的是pearson相关系数。但是相关系数的计算在样本量很小的时候是很不稳定和准确的。一般来说相关性会在十几个配对样本数目的时候会比较准确,但是ATAC-seq和RNA-seq的十几个配对数目就意味着你要总共要做接近上百的样本。这个数据量对于绝大部分实验设计来说都是不可行的。很多时候远端的peak并不一定会跟基因表达呈现相关性,而是跟基因周围的promoter相关。同时,几个远端的peak还会一起呈现同步变化。
enhancerPromterCor并不接受mmAnno module产生的注释结果,其原理是首先会根据你设置的up_scanPromoter和down_scanPromoter参数来产生一个TSS周围的scan范围,从中找到最近的那个peak,从而定义为对应基因的promoter。然后再根据你设置的up_scanEnhancer和down_scanEnhacner来再次产生scan范围,从而寻找跟你gene link的那些peak。
enhancerPromterCor只需要用户提供peakScoreMt,其会根据上一步建立的distal _peak-promoter link关系来建立相关性。这样的好处是能够用来计算相关性的样本数目变多了,之前是配对的7个时间点,这里我们可以用到16个样本。
mmAnnoCor_ePLink <- enhancerPromoterCor(peak_GR = peak_GR,
Txdb = Txdb,
up_scanPromoter = 500,
down_scanPromoter = 500,
up_scanEnhancer = 2e4,
down_scanEnhacner = 2e4,
peakScoreMt = ATAC_normCount)
## >> checking seqlevels match... 2021-08-16 19:20:48
## >> your peak_GR seqlevel:Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## >> using scanPromoter parameter to scan promoter for each gene... 2021-08-16 19:20:48
## >> checking seqlevels match... 2021-08-16 19:20:48
## >> your peak_GR seqlevel:Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## >> some scan range may cross Chr bound, trimming... 2021-08-16 19:20:49
## >> there are 85 gene have scaned promoter
## >> using scanEnhancer parameter to scan Enhancer for each gene... 2021-08-16 19:20:49
## >> checking seqlevels match... 2021-08-16 19:20:49
## >> your peak_GR seqlevel:Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## >> some scan range may cross Chr bound, trimming... 2021-08-16 19:20:50
## >> calculating cor and pvalue, which may be time consuming... 2021-08-16 19:20:50
## >> merging all info together... 2021-08-16 19:20:50
## >> done 2021-08-16 19:20:50可以看到,相比于前面peakGeneCor的结果,enhancerPromoterCor的结果还多了一列promoter_feature,这一列就标明了gene_id那一列对应基因的promoter。
mmAnnoCor_ePLink
## GRanges object with 836 ranges and 8 metadata columns:
## seqnames ranges strand | feature_id gene_id
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] Chr5 27-311 * | WFX_peak_19629 AT5G01010
## [2] Chr5 501-2153 * | WFX_peak_19630 AT5G01010
## [3] Chr5 2611-2965 * | WFX_peak_19631 AT5G01010
## [4] Chr5 8093-10183 * | WFX_peak_19633 AT5G01010
## [5] Chr5 14186-14516 * | WFX_peak_19634 AT5G01010
## ... ... ... ... . ... ...
## [832] Chr5 345002-345964 * | WFX_peak_19723 AT5G01920
## [833] Chr5 346146-346877 * | WFX_peak_19724 AT5G01920
## [834] Chr5 348523-348794 * | WFX_peak_19725 AT5G01920
## [835] Chr5 349128-349354 * | WFX_peak_19726 AT5G01920
## [836] Chr5 357097-358081 * | WFX_peak_19727 AT5G01920
## distanceToTSS promoter_feature cor pvalue padj qvalue
## <numeric> <character> <numeric> <numeric> <numeric> <numeric>
## [1] 4749 WFX_peak_19632 0.625633 0.00953818 1 0.0456379
## [2] 2907 WFX_peak_19632 0.586540 0.01693224 1 0.0570229
## [3] 2095 WFX_peak_19632 0.619536 0.01048138 1 0.0463258
## [4] -3031 WFX_peak_19632 0.536575 0.03212319 1 0.0859153
## [5] -9124 WFX_peak_19632 0.427537 0.09856432 1 0.1745299
## ... ... ... ... ... ... ...
## [832] -13142 WFX_peak_19728 -0.095896 0.7238754 1 0.536263
## [833] -12229 WFX_peak_19728 -0.442621 0.0860029 1 0.164300
## [834] -10312 WFX_peak_19728 -0.116823 0.6665642 1 0.527060
## [835] -9752 WFX_peak_19728 -0.440177 0.0879562 1 0.165932
## [836] -1025 WFX_peak_19728 0.153407 0.5705587 1 0.486139
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths如果用户想要提取出gene_id和promoter的对应关系,可以用
metadata(mmAnnoCor_ePLink)$mm_promoter_tidy
## # A tibble: 85 x 3
## gene_id distanceToTSS promoter_feature
## <chr> <dbl> <chr>
## 1 AT5G01010 0 WFX_peak_19632
## 2 AT5G01015 279 WFX_peak_19632
## 3 AT5G01020 0 WFX_peak_19633
## 4 AT5G01030 0 WFX_peak_19633
## 5 AT5G01040 0 WFX_peak_19635
## 6 AT5G01050 0 WFX_peak_19637
## 7 AT5G01070 -257 WFX_peak_19638
## 8 AT5G01075 -41 WFX_peak_19639
## 9 AT5G01090 0 WFX_peak_19642
## 10 AT5G01100 0 WFX_peak_19644
## # ... with 75 more rows就如同前面peakGeneCor一样,enhancerPromoterCor的结果也可以用plot_peakGeneCor和shinyParse_peakGeneCor进行展示和探究。只不过这里不再是基因和其周围peak的相关性散点图,而是基因promoter和其distal peak的。
plot_peakGeneCor(mmAnnoCor = mmAnnoCor_ePLink,
select_gene = "AT5G01010")
## Joining, by = "feature_id"
## Joining, by = "feature_id"
## `geom_smooth()` using formula 'y ~ x'
shinyParse_peakGeneCor(mmAnnoCor = mmAnnoCor_ePLink)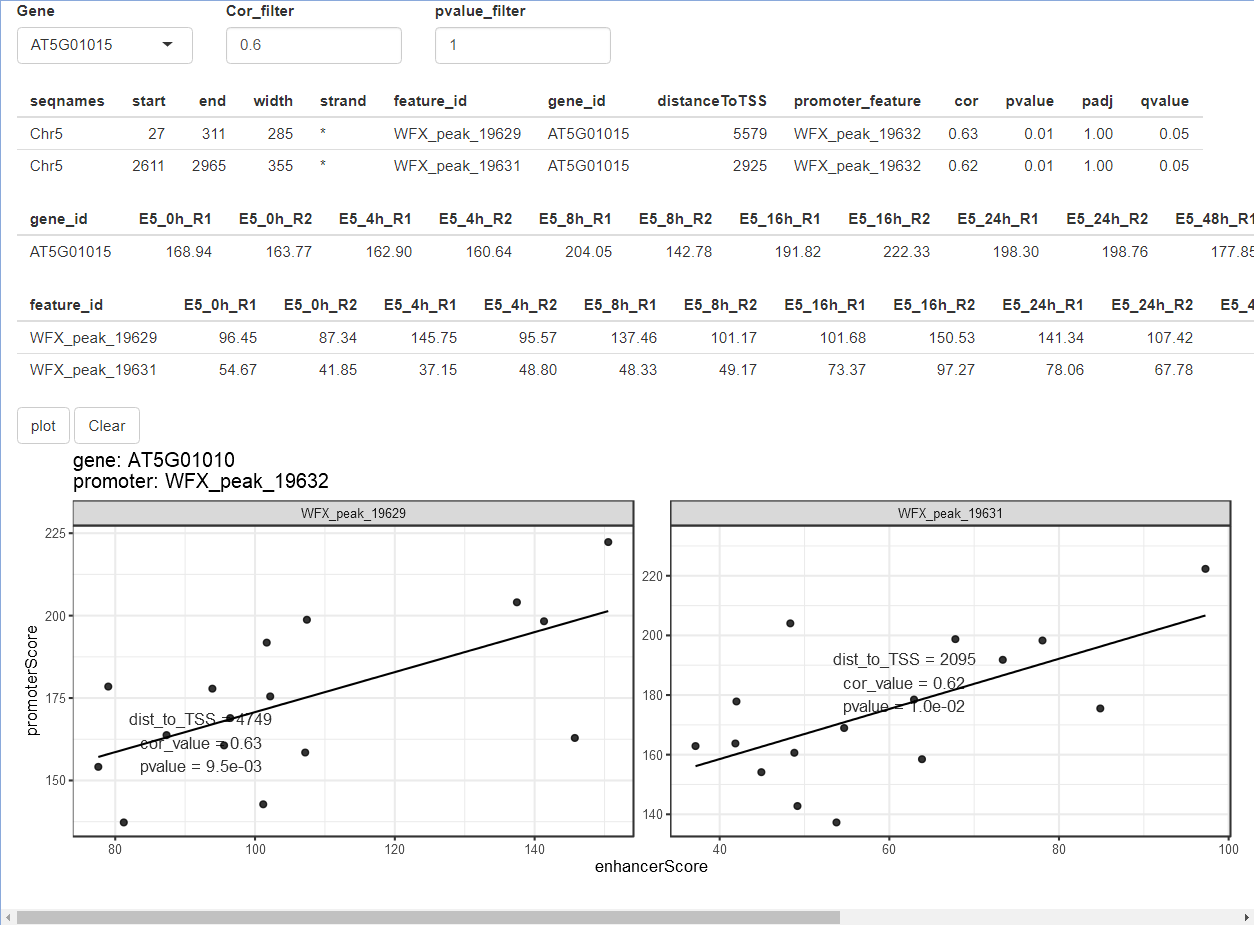
Yoshida, H., Lareau, C.A., Ramirez, R.N., Rose, S.A., Maier, B., Wroblewska, A., Desland, F., Chudnovskiy, A., Mortha, A., Dominguez, C., et al. (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897-912.e20.↩︎
Corces, M.R., Granja, J.M., Shams, S., Louie, B.H., Seoane, J.A., Zhou, W., Silva, T.C., Groeneveld, C., Wong, C.K., Cho, S.W., et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898.↩︎
Guandong Shang
PhD Candidate
My research interests include rstats, epigenomics, bioinformatics and evo-devo.