Bioconductor包投递指南
Bioconductor和CRAN,都是一个可以接受R包的地方。相比于后者而言,Bioconductor更像一个专门针对生物信息学和计算生物学的社群。这使得投递R包到Bioconductor更加的困难,这一点从三个网站的R包数目就可以看出来。
# https://stackoverflow.com/questions/63240125/get-the-current-numbers-of-cran-packages-and-bioconductor-packages
> Sys.time()
[1] "2021-08-06 08:42:56 CST"
# CRAN
> nrow(available.packages())
[1] 17942
# Bioconductor
> library(rvest)
> url <- 'https://www.bioconductor.org/packages/devel/bioc/'
> biocPackages <- url %>% read_html() %>% html_table() %>%.[[1]]
> nrow(biocPackages)
[1] 2015
但这里的困难并不是说Bioconductor的R包的结构,语法等要比CRAN的R包要难,而是说Bioconductor对R包的投递要求更多,更加地复杂。这也使得很多有心投递R包到Bioconductor的人望而却步。考虑到国内缺乏一个比较完善的R包投递指南,我在这里就根据自己的投递经验写一下针对Bioconductor的投递指南,希望能够帮助更多的人参与到Bioconductor社群建设中。
事先声明下,这篇文章并不是教你如何写R包,而是如何教你跳过一些坑,从而相对顺利地投递R包。对于R包的开发,还是推荐hadley wickham的《R packages》1
对于Bioconductor包的开发和投递,除了上面提到的《R packages》之外,最好的资源就是Bioconductor自己官网上的指南了。
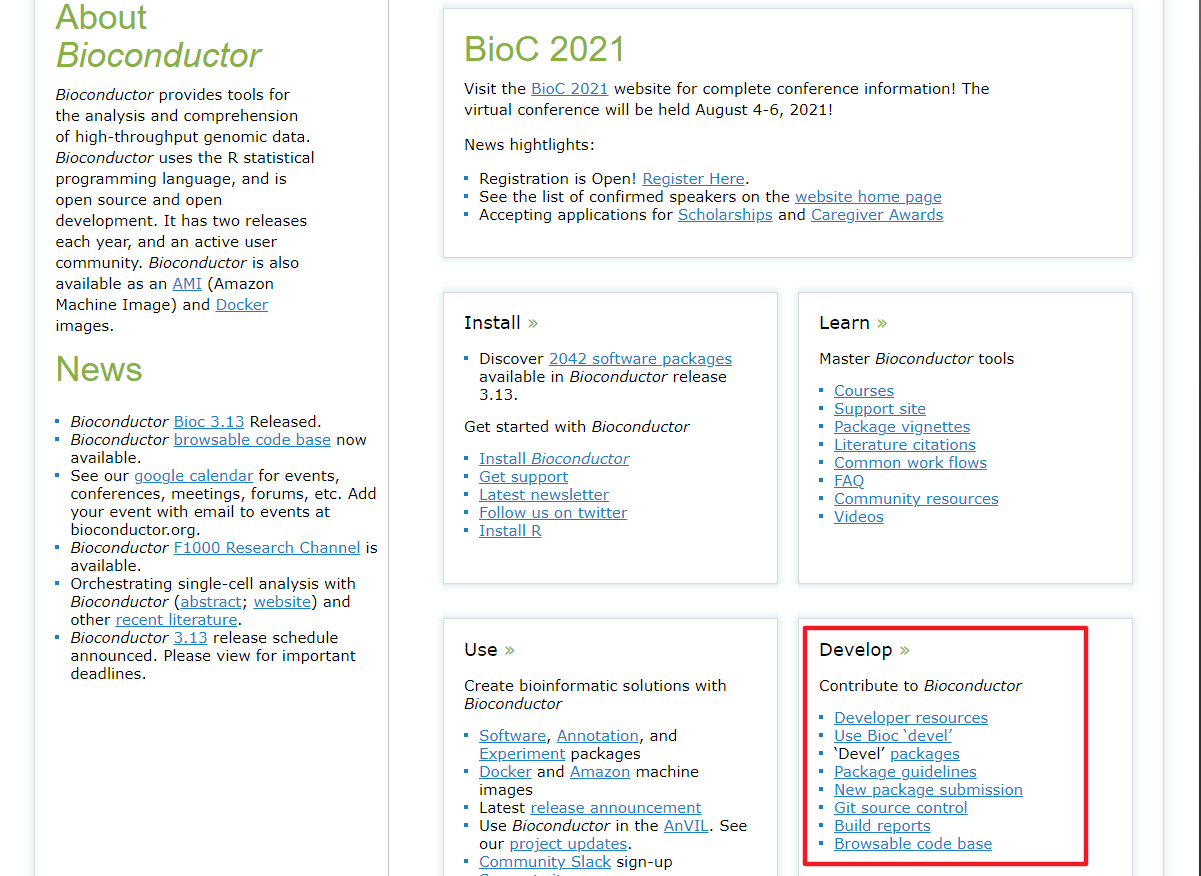 这其中
这其中Developer resources这一栏对于刚入门的开发者而言是最重要的,里面包含了Bioconductor的R包的代码格式,结构语法,版本控制要求等指南,这些都是在开放和投递的时候需要你反复地查看的。对于还在R包开放阶段或者有志于R包开放的人来说,首先应该看的是这一栏里面的Bioconductor Package Guidelines for Developers and Reviewers和Package Submission。对于这里面的内容我就不逐字翻译了,在这里我着重提一些我认为在R包开放阶段比较重要或者容易踩坑的地方。
- The DESCRIPTION file
DESCRIPTION file里面描述了你R包的很多基本信息,包括你R包的名字和基本功能,作者的名字,版本号,还有你R包的依赖包等。这其中有几个坑需要避免,首先就是R包名字的问题。
This should match the GitHub repository name and is case-sensitive. A package name should be descriptive and should not already exist as a current package (case-insensitive) in Bioconductor nor CRAN. Avoid names that are easily confused with existing package names, or that imply a temporal (e.g., ExistingPackage2) or qualitative (e.g., ExistingPackagePlus) relationship.
对于投递到Bioc的R包,名字不能和已有的R包或者CRAN上的R包有名字上的冲突和歧义,不管是大小写还是其他的什么。我就踩到了这个坑上,我的R包名字叫做FindIT2,是Find Infulential TF and Target的缩写。但关键是在CRAN上有个包叫FindIt,这就明显地上面不符合Bioc的要求。这一点在我的R包review阶段就被提了出来,幸好我可能是第一个犯这个错误的,review就绕过了我。
事实上,上面那段要求也是在我的R包投递之后才堪堪加上的╮(╯_╰)╭
除了R包的名字,版本号也是个大坑。如果你注意看Bioconductor submit的github issue里面,被closed的很多R包都是由于version number不对导致的。
All Bioconductor packages use an x.y.z version scheme. See Version Numbering for specifics to how the release and devel Bioconductor versioning proceeds. When first submitted to Bioconductor, a package should have pre-release version 0.99.0.
对于投递到Bioconductor的github上的R包,其版本号必须是0.99.0,如果是其他的数字,Bioconductor bot就会直接closed掉你的issue。
Author这一栏也有很多值得注意的地方,在R包投递的check过程中,尤其会重点检查Maintainer的email地址。这个email地址必须已经被登记在Bioconductor support和emailing list上。
Depends, Imports, Suggests这三栏可以说是Bioconductor包开发和投递最重要的一项了。Bioc对于投递的R包最基本的要求就是尽可能地使用已经有的成熟method和class。
All packages must be available via Bioconductor or CRAN; the use of the Remotes: field is not supported, hence dependencies only available on other repositories (e.g. GitHub) are not allowed.
Reuse, rather than re-implement or duplicate, well-tested functionality from other packages. Make use of appropriate existing packages (e.g., biomaRt, AnnotationDbi, Biostrings) and classes (e.g., SummarizedExperiment, GRanges, Rle, DNAStringSet), and avoid duplication of functionality available in other Bioconductor packages. See Common Bioconductor Methods and Classes. Bioconductor reviewers are very strict on this point! New packages should be interoperable with existing Bioconductor classes and should not reimplement functionality especially with regards to importing/reading data.
毕竟投递到Bio上的包类型主要是跟生物信息学或者计算生物学相关,而Bioc自己的一些核心class和method已经可以满足绝大部分的分析目的了。对于重复造轮子的R包肯定会在review阶段被要求修改的。还有一点Bioc在这个手册没有提到的是,Bioc似乎更加地偏好S4对象,这一点大家在写自己类的时候可以注意下。
- Documentation
document这里并没有什么特别的坑要说,只是想提下vignette还是要写的仔细点的,这既是对自己负责也是对用户负责。如果写的太过于粗糙,reviewer也会提出来要你改进。在写Bioc相关的vignette时候,可以考虑使用BiocStyle这个Bioc专门的format style包来写。
- Unit tests
unit test是一定要写的,如果你不写的话,reviewer肯定是会要你补的。一开始我非常的抗拒写unit test,因为感觉这玩意很复杂,对我来说有点难以接受。但尝试着写了几个简单的之后,不得不感慨真香。unit test一个很大的作用就是让你在修改代码的时候没有后顾之忧,如果你修改的代码跟之前的结果不一致,那么在check的时候unit test就会报error,这会使得你重新检查你的代码,从而让结果具有可重复性。
- The .gitignore file
.gitignore 这里只有一个坑要避免,Bioconductor不希望.rproj出现在你的github仓库中。但是devtools在use_git()的时候只会把.rproj写到.Rbuildignore里面,而不会把这个写到.gitignore中。换而言之,这个忽略语法得你自己加进去,你如果自己不写进去的话,push的时候,Rstudio附带的Rroject文件还是会在你的Github仓库中。
在Bioconductor包的开发中,除了普通R包开发经常要使用的命令load_all, document, check以外,还需要反复使用一个Bioconductor R包特有的check命令,BiocCheck。这个命令属于同名的BiocCheck包。这个命令除了检查一些简单语法之外,还会检查你的R包中的信息符不符合Bioconductor自身的要求。经常性的使用BiocCheck可以帮助你跳过大部分坑。除了BiocCheck之外,还有一个非官方的Bioc包,biocthis,也非常有用,他会给出一些建议和模版帮助你更好地修改你的R包。
同时,在R包的开发和投递的过程中,如果有什么不懂的地方,在Google无果之外,可以考虑mailing list和slack这两个渠道进行询问。
在完成了你初步的R包开发之后,就可投递到Bioconductor的github仓库了。所谓的投递,其实就是开一个issue,写上你的github仓库名字以及勾选一些选项。在投递前,请再次详细地阅读这个github仓库的README,避免一些不必要的踩坑。在开完issue以及把一个SSH public key加入到你的Github账户之后,就会立刻有个github bot来告诉你一些相关信息,如果你的DESCRIPTION文件对的话,就会加一个1. awaiting moderation label 到你的issue。
在大约3天之后,Bioconductor就会分配给你一个reviewer,同时加入2. review in progress label。这时候你的R包就会进入build的阶段,Bioconductor会在三个平台(Linux,Mac和Win)检验你的R包是否能够运行成功。如果不成功的话,就会再加EORROR或者WARNING label。你必须修改你的R包,直到不报错为止,这样你才能进入下一个阶段。这时候你就需要在本地修改,然后分别把本地的修改版本push到github和Bioconductor的仓库。需要注意的一点是,你每次做了修改,然后决定push的时候, 必须bump version,即version number从原来的0.99.0到0.99.1。具体的push等操作可以查看Bioconductor自己的指南2. 我在这里踩了个小坑,就是rtracklayer::import.bw()函数在windows的32位R中运行会报错,这使得我的R包在build的时候就报了error。这里有一个小tips可以帮助你尽可能地减少error的概率,就是偶尔看看别人的package issue,因为上面会有详细的build report,这样你就可以知道要避免哪些错误了。
在build成功之后,OK label就会被加入,这时候reviewer就会从多个角度开始审核你的R包了,包括DESCRIPTION,NAMESPACE,vignettes,R,test等。大约1周之后就会给出详细的reviewer意见,这时候你就要根据reviewer的意见进行修改。修改完成之后,然后再@ reviewer,告诉他你已经完成了修改。经过多轮的反复修改和讨论,最后他会将3a. accepted label加入。然后你的R包就可以正式加入到Bioconductor的R包仓库中了。
参考
Guandong Shang
PhD Candidate
My research interests include rstats, epigenomics, bioinformatics and evo-devo.