FindIT2:An R/Bioconductor package to identify influential TF and target
最近开发了一个R包,FindIT2,可以用来对表观数据进行一些综合性的分析,包括peak注释,TF target的寻找,上游TF的发现等等。我个人感觉这个包对于ATAC-seq得到的相关数据尤其有用,同时由于这个包的核心method和classes都是基于Bioconductor的GenomicRanges写的,其对于非模式物种的数据也非常的友好。
FindIT2现在已经提交到了Bioconductor上面: https://bioconductor.org/packages/devel/bioc/html/FindIT2.html 。不过由于Bioconductor自身的版本要求,所以如果要下载Bioconductor上的版本的话,R语言的版本必须要大于4.10。对于R版本不能随便升级的用户来说(比如说日常操作都在服务器上的用户),可以考虑下载我github上的版本: https://github.com/shangguandong1996/FindIT2 。直接使用下面命令即可安装
BiocManager::install("shangguandong1996/FindIT2")
FindIT2主要由5个核心模块组成,包括了两个元件注释模块,一个调控潜能计算模块,一个TF靶基因计算模块和一个上游TF探究模块。
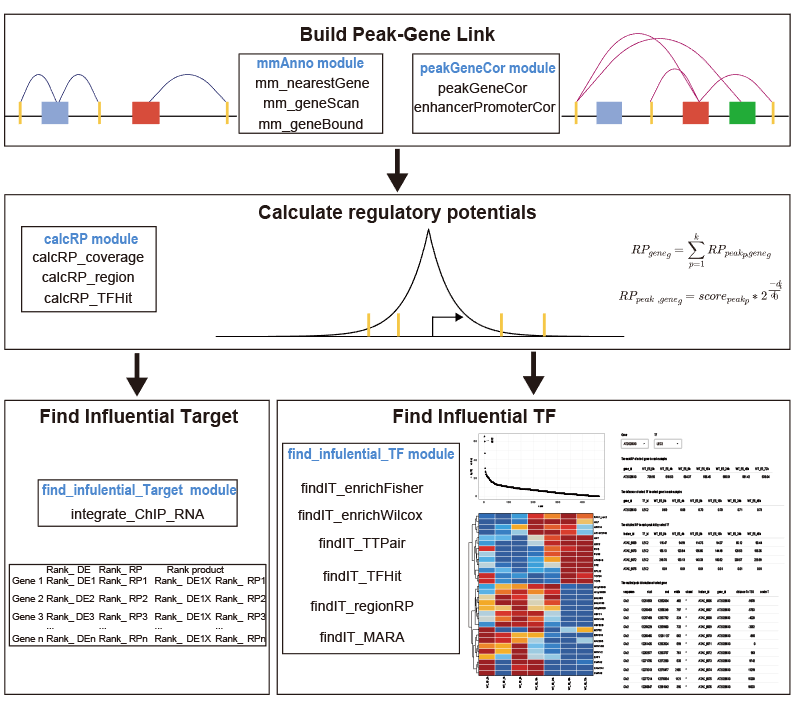 每个模块中还有根据物种注释信息和实验设计的不同来行驶不同功能的子函数。同时FindIT2还有其他的一些辅助函数来帮助用户更好地探究结果。具体的每个模块及内部函数的解读我会在后面的文章中一一解释,也可以查看包的文档:
https://bioconductor.org/packages/devel/bioc/vignettes/FindIT2/inst/doc/FindIT2.html 。在这篇文章中我主要是大致描述下每个模块的功能。
每个模块中还有根据物种注释信息和实验设计的不同来行驶不同功能的子函数。同时FindIT2还有其他的一些辅助函数来帮助用户更好地探究结果。具体的每个模块及内部函数的解读我会在后面的文章中一一解释,也可以查看包的文档:
https://bioconductor.org/packages/devel/bioc/vignettes/FindIT2/inst/doc/FindIT2.html 。在这篇文章中我主要是大致描述下每个模块的功能。
mmAnnomodule跟peakGeneCormodule的目的都是将peak和其调控基因所关联起来。不同的是前者仅仅是基于基因组上的位置信息来进行关联,而后者则考虑到了peak与基因之间或者peak与peak之间的动态相关性。mmAnno下面有三个函数,分别是mm_nearestGene,mm_geneScan以及mm_geneBound。这三个函数的应用场景有略微的不同。mm_nearestGene主要基于最近基因注释原则来将peak关联到基因上,从而产生一对一的peak-gene关系,而mm_geneScan则支持多对多的peak-gene关系,这一点在你的peak数目比较少,而你又不想忽略一些重要基因的时候尤其有用。前两个函数都是为peak来寻找peak-gene的link,而mm_geneBound则是用来为你的输入基因来寻找相关的peak,这个应用场景主要在画peak的火山图或者热图的时候。peakGeneCor模块则是在前面mmAnno模块的注释结果上,根据peak和基因的表达量结果来计算相关性,从而帮助用户找到更加相关的peak-gene link结果。具体的使用方法可以参见包的文档或者后续的文章。
calcRP模块的构思主要来自于刘小乐实验室的一些文章1。该模块下属也有三个函数:calcRP_coverage主要是通过bigwig文件来计算调控潜能,calcRP_regionRP主要是通过peakCount矩阵以及peak-gene link来计算调控潜能,而calcRP_TFHit则是基于ChIP-seq的call peak结果。前两个函数的输入对象都是ATAC-seq的结果,目的主要是将每个基因周围的染色质状态合并成一个分数来方便后续的一些计算。而第三个函数则是将每个基因周围的TF ChIP-seq的靶位点合并成一个分数,从而产生一个排序结果,方便用户筛选靶基因。这个函数跟刘小乐实验室之前的BETA2软件非常相似,但相比于BETA只支持人和小鼠的数据,该函数支持任何含有基因组注释的物种,这一点对于非模式物种非常的有帮助。
find_influential_Target模块只有一个函数:integrate_ChIP_RNA。该函数将前面calcRP_TFHit的结果作为输入,同时可以整合TF过表达/突变的RNA-seq数据,来更加精确地找到TF的靶基因。前面calcRP_TFHit实现的是BETA的minus模块,而这里的联合分析则实现的是BETA的Basic模块。
find_influential_TF模块中有多个函数。其中findIT_TFHit,findIT_regionRP,findIT_TTpair可以为感兴趣的基因找到其潜在的上游TF。而findIT_enrichWilcox,findIT_enrichFisher,findIT_MARA则可以为peak找到其潜在的上游TF。同时各个函数又有其他的输入数据要求,这些我都会在后续的文章中提到。再一次的,得益于Bioconductor的method和class,这其中大部分函数对非模式物种都是非常友好的。
总的来说,作为一个用了很多包和分析了很多数据的用户,我个人感觉这个包总体来说还是很有用的,尤其是对那些注释不完善的非模式物种而言。欢迎大家多多测试并提出issue。
参考:
Guandong Shang
PhD Candidate
My research interests include rstats, epigenomics, bioinformatics and evo-devo.