Intro_FindIT2:find_influential_TF module part1
从这篇文章开始,我们进入FindIT2的最后一个模块:find_influential_TF module,即寻找调控你感兴趣peak或者基因的那些TF。由于这个模块里面的各个函数的基本思想大不相同,所以我会将这个模块的介绍拆分成几篇文章。在这篇文章中我介绍的是如何寻找调控你感兴趣基因的那些TF,感兴趣的基因集可以来自于差异分析、聚类等等分析结果。这部分包含了两个函数:findIT_TTPair和findIT_TFHit。前者基于的是Fisher精确检验,后者基于的是Wilcox秩和检验,具体的细节我们会在后面讨论。
Pre-preparation
这次的测试数据有些复杂,包含了LEC2的在整个基因组上的ChIP-seq结果,GR诱导LEC2过表达的RNA-seq结果以及一个ATAC的多时间点的mergePeak文件以及一些公共数据。
# 加载包
library(FindIT2)
## Warning: package 'GenomeInfoDb' was built under R version 4.1.1
library(TxDb.Athaliana.BioMart.plantsmart28)
## Warning: package 'GenomicFeatures' was built under R version 4.1.1
library(BSgenome.Athaliana.TAIR.TAIR9)
library(motifmatchr)
library(dplyr)
library(ggplot2)
Txdb <- TxDb.Athaliana.BioMart.plantsmart28
seqlevels(Txdb) <- paste0("Chr", c(1:5, "M", "C"))# 下载LEC2在整个基因组上的ChIP-seq结果
download.file(url = "https://raw.githubusercontent.com/shangguandong1996/FindIT2_paper_relatedCode/master/rawdata/ChIP/TF_ChIP_bindingSite.bed",
destfile = "TF_ChIP_bindingSite.bed")
# 下载ATAC peak
download.file(url = "https://raw.githubusercontent.com/shangguandong1996/FindIT2_paper_relatedCode/master/rawdata/ATAC/ATAC_mergePeak.bed",
destfile = "ATAC_mergePeak.bed")
# 下载RNA-seq的差异分析结果
download.file(url = "https://raw.githubusercontent.com/shangguandong1996/FindIT2_paper_relatedCode/master/rawdata/RNA/LEC2_RNA_diffResult.rda",
destfile = "LEC2_RNA_diffResult.rda")
# 下载公共数据库中的TF-target 配对文件
download.file(url = "http://bioinformatics.psb.ugent.be/webtools/iGRN/files/igrn_data/iGRN_network_full.txt",
destfile = "iGRN_network_full.txt")
# 下载公共数据库中各种TF ChIP-seq的peak文件
download.file(url = "https://remap.univ-amu.fr/storage/remap2022/tair10/tf/MACS2/remap2022_all_macs2_TAIR10_v1_0.bed.gz",
destfile = "remap2022_all_macs2_TAIR10_v1_0.bed.gz")
# 下载公共数据中的Genome-wide TFBS文件
download.file(url = "http://plantregmap.gao-lab.org/download_ftp.php?filepath=08-download/Arabidopsis_thaliana/binding/TFBS_from_motif_genome-wide_Ath.gff",
destfile = "TFBS_from_motif_genome-wide_Ath.gff")
ChIP_peak_GR <- loadPeakFile("TF_ChIP_bindingSite.bed")
ChIP_peak_GR
## GRanges object with 3223 ranges and 2 metadata columns:
## seqnames ranges strand | feature_id score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] Chr1 37901-38160 * | TF_ChIP_1 0.747831
## [2] Chr1 51301-51660 * | TF_ChIP_2 1.368107
## [3] Chr1 51801-51810 * | TF_ChIP_3 0.765108
## [4] Chr1 68451-68660 * | TF_ChIP_4 0.797340
## [5] Chr1 116401-116660 * | TF_ChIP_5 0.853717
## ... ... ... ... . ... ...
## [3219] Chr5 26903801-26904210 * | TF_ChIP_3219 0.829693
## [3220] Chr5 26905801-26906010 * | TF_ChIP_3220 1.008028
## [3221] Chr5 26906401-26906760 * | TF_ChIP_3221 0.990938
## [3222] Chr5 26933051-26933410 * | TF_ChIP_3222 0.814937
## [3223] Chr5 26933851-26934110 * | TF_ChIP_3223 0.943218
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsload("LEC2_RNA_diffResult.rda")
diff_RNA
## # A tibble: 37,051 x 3
## gene_id log2FoldChange padj
## <chr> <dbl> <dbl>
## 1 AT1G01010 -0.0169 0.510
## 2 AT1G01020 0.000174 1.00
## 3 AT1G03987 -0.478 0.191
## 4 AT1G01030 -0.00859 1.00
## 5 AT1G01040 -0.000254 1.00
## 6 AT1G03993 0 NA
## 7 AT1G01050 -0.000576 1.00
## 8 AT1G03997 0 NA
## 9 AT1G01060 -0.00174 1.00
## 10 AT1G01070 -0.00384 1.00
## # ... with 37,041 more rowsATAC_peak_GR <- loadPeakFile("ATAC_mergePeak.bed")
ATAC_peak_GR
## GRanges object with 22994 ranges and 2 metadata columns:
## seqnames ranges strand | feature_id score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] Chr1 30-875 * | ATAC_1 0
## [2] Chr1 1392-1953 * | ATAC_2 0
## [3] Chr1 2164-3625 * | ATAC_3 0
## [4] Chr1 8268-8966 * | ATAC_4 0
## [5] Chr1 13654-14698 * | ATAC_5 0
## ... ... ... ... . ... ...
## [22990] Chr5 26969184-26969915 * | ATAC_22990 0
## [22991] Chr5 26970542-26971504 * | ATAC_22991 0
## [22992] Chr5 26972304-26973186 * | ATAC_22992 0
## [22993] Chr5 26974280-26974825 * | ATAC_22993 0
## [22994] Chr5 26975157-26975438 * | ATAC_22994 0
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengths# 公共数据库中的拟南芥 TF-target的配对文件
TF_target <- readr::read_delim(file = "iGRN_network_full.txt",
delim = "\t")
## Rows: 1709871 Columns: 5
## -- Column specification --------------------------------------------------------
## Delimiter: "\t"
## chr (3): #TF, target_gene, input_networks
## dbl (2): score, rank
##
## i Use `spec()` to retrieve the full column specification for this data.
## i Specify the column types or set `show_col_types = FALSE` to quiet this message.
TF_target
## Warning: One or more parsing issues, see `problems()` for details
## # A tibble: 1,709,871 x 5
## `#TF` target_gene score rank input_networks
## <chr> <chr> <dbl> <dbl> <chr>
## 1 AT3G54340 AT3G09620 0.993 1 "\tCMM\t\t\tChIP\tCOE\tGENIE3\t"
## 2 AT3G54340 AT2G42830 0.992 2 "\tCMM\t\t\tChIP\tCOE\tGENIE3\t"
## 3 AT5G20240 AT1G69120 0.992 3 "\tCMM\t\t\tChIP\tCOE\tGENIE3\t"
## 4 AT3G59060 AT1G03600 0.992 4 "PWM\tCMM\tDH\t\tChIP\tCOE\tGENIE3\t"
## 5 AT2G43010 AT4G04330 0.991 5 "PWM\tCMM\tDH\t\tChIP\tCOE\tGENIE3\t"
## 6 AT3G54340 AT3G02310 0.990 6 "\tCMM\t\t\tChIP\tCOE\tGENIE3\t"
## 7 AT5G13790 AT1G27930 0.990 7 "PWM\tCMM\tDH\t\tChIP\tCOE\tGENIE3\t"
## 8 AT2G43010 AT2G45740 0.989 8 "PWM\t\tDH\t\tChIP\tCOE\tGENIE3\t"
## 9 AT2G43010 AT3G26180 0.989 9 "PWM\t\t\t\tChIP\tCOE\tGENIE3\t"
## 10 AT2G43010 AT5G11060 0.989 10 "PWM\tCMM\tDH\t\tChIP\tCOE\tGENIE3\t"
## # ... with 1,709,861 more rows# Remap2020的bed文件
database_ChIP_GR <- loadPeakFile("remap2022_all_macs2_TAIR10_v1_0.bed.gz")
database_ChIP_GR
## GRanges object with 4843030 ranges and 4 metadata columns:
## seqnames ranges strand | feature_id
## <Rle> <IRanges> <Rle> | <character>
## [1] 1 1-1033 * | GSE143831.MED12.Col-..
## [2] 1 1-1053 * | GSE124750.AGO4.Col_i..
## [3] 1 1-1194 * | GSE124750.AGO6.Col_i..
## [4] 1 1-1198 * | GSE124750.AGO9.Col_i..
## [5] 1 1-1234 * | GSE124750.AGO9.Col_i..
## ... ... ... ... . ...
## [4843026] 5 26975133-26975442 * | GSE166543.EICBP-B.Co..
## [4843027] 5 26975155-26975443 * | GSE124750.AGO4.Col_i..
## [4843028] 5 26975174-26975444 * | GSE108673.SPT6.Col-0..
## [4843029] 5 26975211-26975325 * | GSE60141.TGA4.Col-0_..
## [4843030] 5 26975213-26975502 * | GSE60141.TRP2.Col-0_..
## score itemRgb thick
## <numeric> <character> <IRanges>
## [1] 4.41114 #8C381C 500
## [2] 17.08726 #674F67 313
## [3] 21.07903 #556C10 294
## [4] 58.29619 #22DBC3 415
## [5] 7.67439 #22DBC3 267
## ... ... ... ...
## [4843026] 6.47935 #CDE461 26975253
## [4843027] 10.07379 #674F67 26975286
## [4843028] 12.09363 #F8D7EE 26975298
## [4843029] 5.75710 #DB9A42 26975291
## [4843030] 355.36154 #F7A31C 26975406
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengths# PlantRegMap的genome-wide motif scan文件
plantTFDB_TFBS_GR <- import.gff("TFBS_from_motif_genome-wide_Ath.gff")
plantTFDB_TFBS_GR
## GRanges object with 5606619 ranges and 7 metadata columns:
## seqnames ranges strand | source type score
## <Rle> <IRanges> <Rle> | <factor> <factor> <numeric>
## [1] Chr1 2-16 + | fimo motif 92.5
## [2] Chr1 4-21 + | fimo motif 50.8
## [3] Chr1 5-27 + | fimo motif 136.7
## [4] Chr1 6-24 + | fimo motif 76.0
## [5] Chr1 9-23 + | fimo motif 92.5
## ... ... ... ... . ... ... ...
## [5606615] ChrM 366758-366778 + | fimo motif 58.8
## [5606616] ChrM 366761-366782 + | fimo motif 82.1
## [5606617] ChrM 366764-366782 - | fimo motif 50.8
## [5606618] ChrM 366764-366782 + | fimo motif 52.9
## [5606619] ChrM 366768-366782 + | fimo motif 50.2
## phase Name ID sequence
## <integer> <character> <character> <character>
## [1] <NA> AT1G72740 AT1G72740_1 CCTAAACCCTAAACC
## [2] <NA> AT2G33550 AT2G33550_2 TAAACCCTAAACCCTAAA
## [3] <NA> AT1G19790 AT1G19790_3 AAACCCTAAACCCTAAACCC..
## [4] <NA> AT5G67580 AT5G67580_4 AACCCTAAACCCTAAACCC
## [5] <NA> AT1G72740 AT1G72740_5 CCTAAACCCTAAACC
## ... ... ... ... ...
## [5606615] <NA> AT3G23250 AT3G23250_5606615 TGCCACCTCCCTCAACAACCT
## [5606616] <NA> AT1G18960 AT1G18960_5606616 CACCTCCCTCAACAACCTGACC
## [5606617] <NA> AT3G47600 AT3G47600_5606617 GGTCAGGTTGTTGAGGGAG
## [5606618] <NA> AT1G08810 AT1G08810_5606618 CTCCCTCAACAACCTGACC
## [5606619] <NA> AT1G09540 AT1G09540_5606619 CTCAACAACCTGACC
## -------
## seqinfo: 7 sequences from an unspecified genome; no seqlengthsintegrate_ChIP_RNA
这篇文章的目的主要是向大家介绍如何用findIT_TTPair和findIT_TFHit来找调控感兴趣基因的TF,所以首先我们需要得到一个感兴趣的基因集。这里我用LEC2的靶基因作为我感兴趣的基因集,从而看这两个函数能否成功地识别出LEC2是这些基因集的调控TF。
这里我用上一篇文章介绍过的integrate_ChIP_RNA函数来得到LEC2的靶基因
# 我希望calcRP_TFHit能够将我bed文件里面的score考虑在内
# 所以我重命名了socre列为featrure_score
colnames(mcols(ChIP_peak_GR))[2] <- "feature_score"
mmAnno_TFHit <- mm_geneScan(ChIP_peak_GR,
Txdb,
upstream = 2e4,
downstream = 2e4)
## >> checking seqlevels match... 2021-09-25 19:50:00
## >> your peak_GR seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## ------------
## annotatePeak using geneScan mode begins
## >> preparing gene features information and scan region... 2021-09-25 19:50:00
## >> some scan range may cross Chr bound, trimming... 2021-09-25 19:50:01
## >> finding overlap peak in gene scan region... 2021-09-25 19:50:01
## >> dealing with left peak not your gene scan region... 2021-09-25 19:50:01
## >> merging two set peaks... 2021-09-25 19:50:01
## >> calculating distance and dealing with gene strand... 2021-09-25 19:50:01
## >> merging all info together ... 2021-09-25 19:50:01
## >> done 2021-09-25 19:50:01
TFHit_RP <- calcRP_TFHit(mmAnno = mmAnno_TFHit,
Txdb = Txdb,
decay_dist = 1000,
report_fullInfo = FALSE)
## >> calculating peakCenter to TSS using peak-gene pair... 2021-09-25 19:50:01
## >> calculating RP using centerToTSS and feature_score 2021-09-25 19:50:02
## >> merging all info together 2021-09-25 19:50:02
## >> done 2021-09-25 19:50:02
merge_data <- integrate_ChIP_RNA(result_geneRP = TFHit_RP,
result_geneDiff = diff_RNA,
lfc_threshold = 0.58,
padj_threshold = 0.05)
# 这里我取前1000个基因作为LEC2的靶基因
LEC2_target <- merge_data$data$gene_id[1:1000]findIT_TTPair
findIT_TTPair的原理跟常见的GO富集分析是一样的,先构建一个列联表,然后进行Fisher精确检验。在GO富集中,想要看的是每个GO条目在感兴趣基因集中是否富集,而在findIT_TTPair中则是看每个TF对应的target gene集合在感兴趣基因集中是否富集。可以想象一下,如果你感兴趣的基因集都是某个TF的target gene,那么自然这个TF就有可能会调控这个基因集。
# 首先我们需要修改下TF_target这个数据框
# 因为findIT_TTPair仅支持一个两列的数据框,且这两列列名分别为TF_id和target_gene
TF_target <- TF_target %>%
select(1:2) %>%
rename(TF_id = `#TF`)
# 然后就可以进行TF的富集分析了
result_TTPair <- findIT_TTPair(input_genes = LEC2_target,
TF_target_database = TF_target)
result_TTPair %>%
as.data.frame() %>%
head(n = 8)
## TF_id num_TFHit_input inputRatio bgRatio pvalue odds_ratio
## 1 AT3G26790 356 356/997 3674/31393 2.312953e-90 4.532247
## 2 AT1G69120 463 463/997 6567/31393 5.599386e-75 3.450452
## 3 AT2G43010 459 459/997 6968/31393 1.953358e-64 3.130755
## 4 AT3G24650 163 163/997 1254/31393 7.318065e-55 5.249190
## 5 AT4G36920 291 291/997 3610/31393 1.115320e-53 3.362733
## 6 AT1G09530 421 421/997 6739/31393 7.564574e-51 2.785516
## 7 AT1G24260 373 373/997 5676/31393 9.101341e-49 2.828265
## 8 AT1G28300 196 196/997 1950/31393 1.444472e-48 3.995636
## padj qvalue rank
## 1 3.275142e-87 8.095337e-88 1
## 2 7.923132e-72 9.798926e-73 2
## 3 2.762048e-61 2.278917e-62 3
## 4 1.034043e-51 6.403307e-53 4
## 5 1.574832e-50 7.807241e-52 5
## 6 1.067361e-47 4.412668e-49 6
## 7 1.283289e-45 4.550671e-47 7
## 8 2.035261e-45 6.319566e-47 8findIT_TTPair的结果包含了多列信息,第1列是TF的id,id的排序是按照p-value来的。p-value越小,这个TF就会在越前面。可以看到LEC2(AT1G28300)在1000多个TF中排名第8,排在它前面还包括了FUS3, AGL7, PIF4, ABI3, AP2, PAP3, AGL9等TF,这些TF或多或少都跟LEC2有关系。
第2列是num_TFHit_input,代表了在你选择的基因集中,有多少基因是对应TF的target。可以看到,在997个select基因中有196个基因是数据库中LEC2(AT1G28300)的target gene。
第3、4列分别是inputRatio和bgRatio。对于LEC2那一列,inputRatio是196/997,代表的是在997个select基因中有196个LEC2的target gene。bgRatio是1950/31393,代表的是在所有31393个基因中,LEC2的target gene有1950个。
大家可能会感到奇怪,明明我选择的是Top1000 gene,但是为什么这里是997。原因就在于有3个LEC2的target基因并不在数据库里面,自然也就不会有了。同样的,拟南芥可能有33000+或者36000+个基因,但是这里却只有31393个基因,原因就是在数据库中只有这些基因。
第5、6列分别是pvalue和odds_ratio,表征了富集结果的显著性。我们可以画个列联表来展示下这两个结果的计算过程。
| TF target | Non-TF target | total | ||
|---|---|---|---|---|
| select gene | 196 | 801 | 997 | |
| left gene | 1754 | 28642 | 320396 | |
| total | 1950 | 29443 | 31393 |
(matrix_fisher <- matrix(c(196, 801, 1754, 28642), nrow = 2, byrow = TRUE))
## [,1] [,2]
## [1,] 196 801
## [2,] 1754 28642
fisher.test(matrix_fisher)
##
## Fisher's Exact Test for Count Data
##
## data: matrix_fisher
## p-value < 2.2e-16
## alternative hypothesis: true odds ratio is not equal to 1
## 95 percent confidence interval:
## 3.375126 4.712768
## sample estimates:
## odds ratio
## 3.995636可能有些人可能会用常见的odds ratio公式去算odds_ratio,然后发现跟这里的结果对不上。
(matrix_fisher[1, 1] / matrix_fisher[2, 1]) / (matrix_fisher[1, 2] / matrix_fisher[2, 2])
## [1] 3.995741原因主要是fisher.test里面对于odds ratio的估计跟常见的公式有些不同,具体的可以参见:Why do odds ratios from formula and R’s fisher.test differ? Which one should one choose?
第7、8列分别是padj和qvalue,都代表了矫正之后的p-value。第9列是rank,rank越小,排名越靠前。
有些用户可能想要用这个结果去画个图来展示富集的结果,尽管FindIT2虽然没有提供一个画图函数,但是大家可以用下面的代码方便地画出点图并自己进行修图:
result_TTPair %>%
tidyr::separate(col = inputRatio,
into = c("TFHit", "input"),
sep = "/") %>%
slice(1:10) %>%
mutate(input = as.numeric(input),
inputRatio = num_TFHit_input / input) %>%
ggplot(aes(x = inputRatio,
y = reorder(TF_id, inputRatio))) +
geom_point(aes(size = odds_ratio,
color = -log10(qvalue))) +
ylab("TF_id")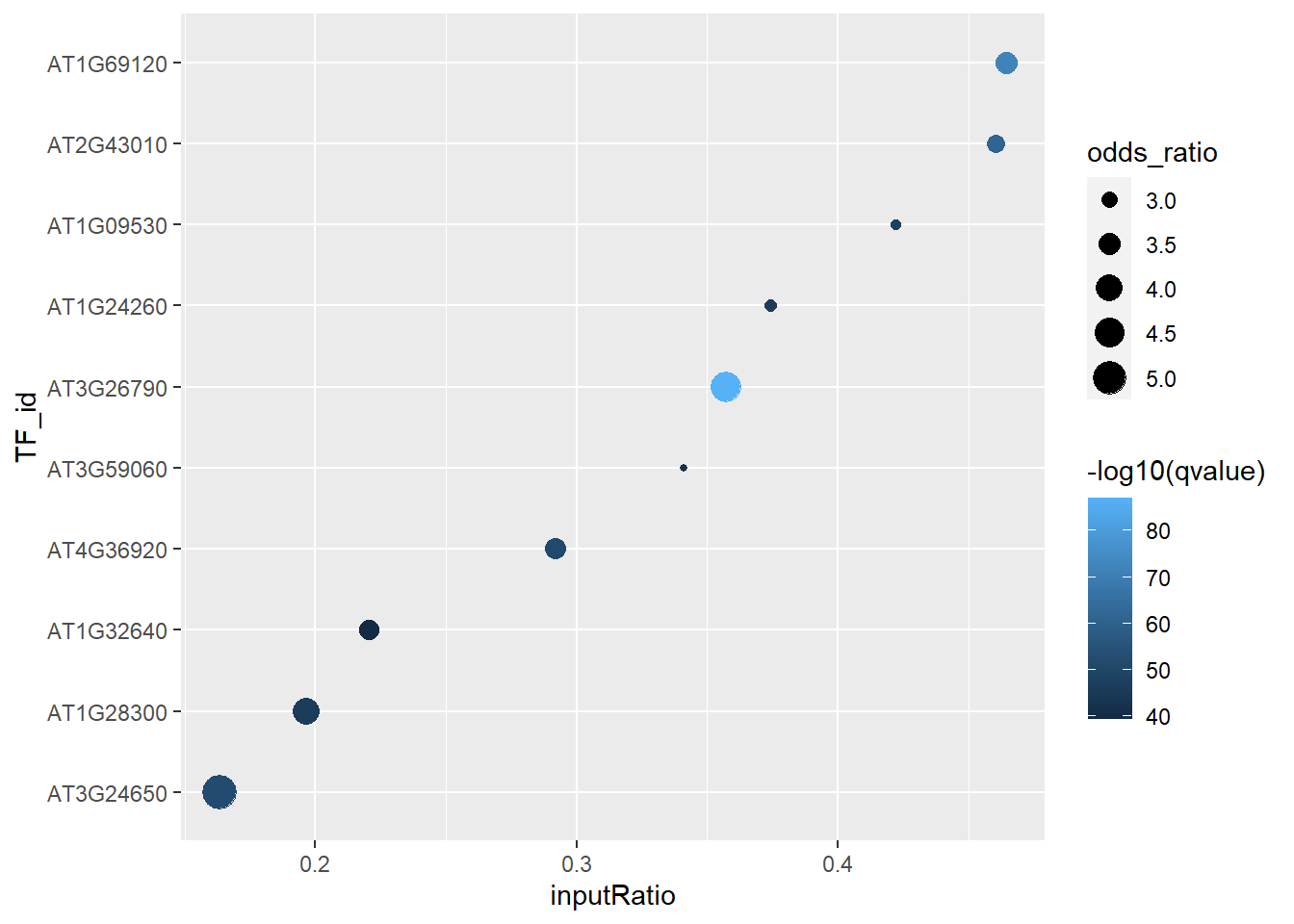
jaccard_findIT_TTpair
在通过findIT_TTPair拿到了在你的基因集中富集的TF之后,我们可能想要看看排名前列的一些TF之间是否存在着关系,比如说在感兴趣的基因集中,这些排名前列TF的target基因是否存在着很大的overlap。为了量化这些overlap,我写了jaccard_findIT_TTpair函数来帮助用户做一个快速地测试。
jaccard_findIT_TTpair基于的是jaccard similarity原理。举个例子,在你的感兴趣基因集中,如果两个TF其并集有300个基因,交集有270个基因,那么其jaccard similarity就是\(270/300=0.9\)。
\[ J(TF_{A}, TF_{B}) = \frac{A \cap B}{A \cup B} \]
# 这里我选择Top10 TF做jaccard计算
result_TTPair_jaccard <- jaccard_findIT_TTpair(input_genes = LEC2_target,
TF_target_database = TF_target,
input_TF_id = result_TTPair$TF_id[1:10])
result_TTPair_jaccard
## AT3G26790 AT1G69120 AT2G43010 AT3G24650 AT4G36920 AT1G09530 AT1G24260
## AT3G26790 0.0000000 0.2836991 0.2977707 0.3007519 0.2394636 0.2885572 0.2612457
## AT1G69120 0.2836991 0.0000000 0.4406250 0.1320072 0.3962963 0.4144000 0.5626168
## AT2G43010 0.2977707 0.4406250 0.0000000 0.1539889 0.4044944 0.5575221 0.4173765
## AT3G24650 0.3007519 0.1320072 0.1539889 0.0000000 0.1293532 0.1564356 0.1355932
## AT4G36920 0.2394636 0.3962963 0.4044944 0.1293532 0.0000000 0.3745174 0.4008439
## AT1G09530 0.2885572 0.4144000 0.5575221 0.1564356 0.3745174 0.0000000 0.3808696
## AT1G24260 0.2612457 0.5626168 0.4173765 0.1355932 0.4008439 0.3808696 0.0000000
## AT1G28300 0.3174224 0.2047532 0.2062615 0.2960289 0.1849148 0.2003891 0.1878914
## AT3G59060 0.2817680 0.3518519 0.5790514 0.1589862 0.4211712 0.5069307 0.3427495
## AT1G32640 0.2229299 0.2601476 0.3971193 0.1641337 0.2775000 0.3354167 0.2835498
## AT1G28300 AT3G59060 AT1G32640
## AT3G26790 0.3174224 0.2817680 0.2229299
## AT1G69120 0.2047532 0.3518519 0.2601476
## AT2G43010 0.2062615 0.5790514 0.3971193
## AT3G24650 0.2960289 0.1589862 0.1641337
## AT4G36920 0.1849148 0.4211712 0.2775000
## AT1G09530 0.2003891 0.5069307 0.3354167
## AT1G24260 0.1878914 0.3427495 0.2835498
## AT1G28300 0.0000000 0.1832230 0.1718310
## AT3G59060 0.1832230 0.0000000 0.3493976
## AT1G32640 0.1718310 0.3493976 0.0000000需要注意的是,所产生的是一个相似性矩阵,数值越高就越相似,说明两个TF的target gene overlap程度很高。但是为了画图方便,我将相似性矩阵的对角线数值,即TF自身的jaccard similarity设置成了0.
我们可以用热图展现
ComplexHeatmap::Heatmap(result_TTPair_jaccard)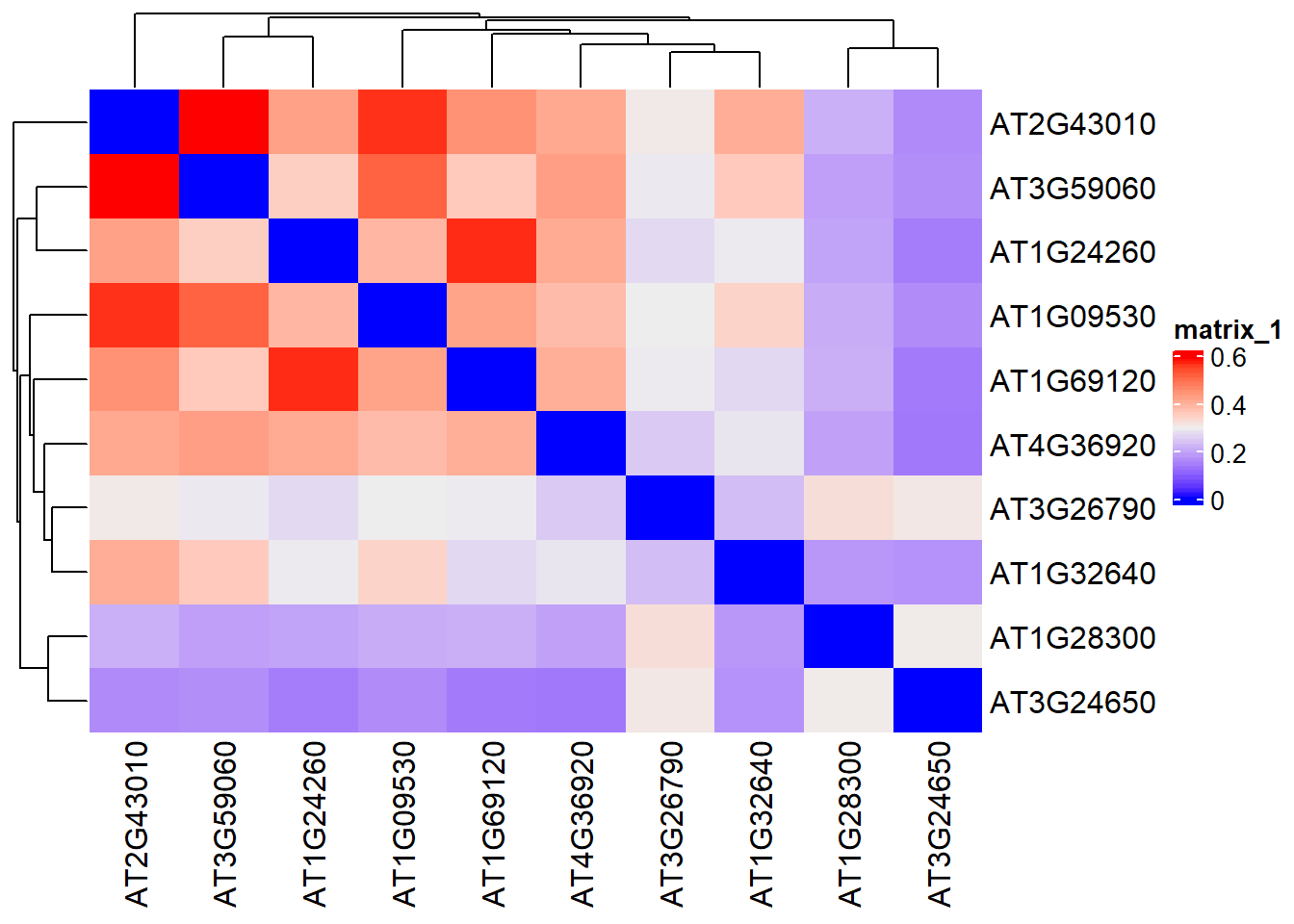
findIT_TFHit
尽管上面的findIT_TTPair原理非常的简单,使用也很方便。但是并不是所有的物种都有如此完善的TF-target数据库,这也就局限了这个函数的应用场景。基于这个原因,我写了findIT_TFHit函数来使得find infulential TF能够扩充到更多的物种。
findIT_TFHit基于的是之前的calcRP_TFHit函数。总的来说,相比于背景基因集,如果一个TF的binding site更多地落在了你感兴趣基因集的附近,那么就说明这个TF很有可能就会是你感兴趣基因集的调控TF。而用于量化某个TF在背景基因集或感兴趣基因集的binding site多少的指标,就是调控潜能分数(regulatory potential,RP)。
为了得到RP,我们就需要知道每个TF的binding site,这里我们用到的是Remap2022数据库中拟南芥各个TF的binding site数据。
# 首先我们需要对数据库的binding site GRange对象做一些修改
# 将seqlevel修改一下
(seqlevels(database_ChIP_GR) <- paste0("Chr", seqlevels(database_ChIP_GR)))
## [1] "Chr1" "Chr2" "Chr3" "Chr4" "Chr5"
# 将feature_id修改成TF_id
colnames(mcols(database_ChIP_GR))[1] <- "TF_id"set.seed(19960203)
result_TFHit <- findIT_TFHit(input_genes = LEC2_target,
Txdb = Txdb,
TF_GR_database = database_ChIP_GR,
scan_dist = 2e4,
decay_dist = 1e3)
## >> preparing gene features information... 2021-09-25 19:50:11
## >> some scan range may cross Chr bound, trimming... 2021-09-25 19:50:12
## >> calculating p-value for each TF, which may be time consuming... 2021-09-25 19:50:14
## >> merging all info together... 2021-09-25 19:51:10
## >> done 2021-09-25 19:51:10
result_TFHit %>%
as.data.frame() %>%
head(n = 10)
## TF_id mean_value TFPeak_number
## 1 GSE145387.VAL1.Col-0_seedling 0.5130745 9754
## 2 GSE145387.VAL2.Col-0_seedling 0.4457701 13039
## 3 GSE26722.REV.Col-0_seedling_10d-DEX-90min 0.3938903 8489
## 4 GSE48081.KAN1.Col-0_seedling_10d-rep1 0.4757618 13815
## 5 GSE94307.HEC1.Col-0_seedling_12d 0.4541106 9082
## 6 GSE142621.BRM.Col-0_seedling_BRIP1 0.3749682 8247
## 7 GSE122579.AT2G17950.Col-0_seedling 0.4619562 10908
## 8 GSE115358.AT2G14045.Col_seedling_TCP4-OE 0.4792326 13926
## 9 GSE80566.AT5G50360.Col-0_seedling_4d-DEX-6h 0.4728792 18597
## 10 GSE52400.BBM.Col-0_somatic-embryo 0.4502585 16348
## pvalue padj qvalue rank
## 1 3.218602e-181 2.233710e-178 4.377299e-179 1
## 2 1.398473e-130 9.691419e-128 9.509618e-129 2
## 3 7.509653e-128 5.196680e-125 3.404376e-126 3
## 4 2.645184e-122 1.827822e-119 8.993626e-121 4
## 5 1.168732e-121 8.064248e-119 3.178950e-120 5
## 6 4.881154e-120 3.363115e-117 1.106395e-118 6
## 7 1.848141e-113 1.271521e-110 3.590675e-112 7
## 8 4.868389e-110 3.344583e-107 8.276260e-109 8
## 9 7.733550e-100 5.305215e-97 1.168625e-98 9
## 10 4.952393e-99 3.392389e-96 6.735255e-98 10可以看到排名前10的并没有出现LEC2,这并不是因为LEC2不显著,而是因为在数据库中并没有LEC2相关的TF ChIP-seq数据收录。我们可以将我们自己的LEC2 ChIP-seq结果加入到数据库里面再跑一次。
mcols(database_ChIP_GR) <- mcols(database_ChIP_GR)[, 1, drop = FALSE]
mcols(ChIP_peak_GR) <- mcols(ChIP_peak_GR)[, 1, drop = FALSE]
ChIP_peak_GR$TF_id <- "LEC2"
database_ChIP_GR <- c(database_ChIP_GR, ChIP_peak_GR)set.seed(19960203)
result_TFHit <- findIT_TFHit(input_genes = LEC2_target,
Txdb = Txdb,
TF_GR_database = database_ChIP_GR,
scan_dist = 2e4,
decay_dist = 1e3)
## >> preparing gene features information... 2021-09-25 19:51:11
## >> some scan range may cross Chr bound, trimming... 2021-09-25 19:51:11
## >> calculating p-value for each TF, which may be time consuming... 2021-09-25 19:51:12
## >> merging all info together... 2021-09-25 19:52:07
## >> done 2021-09-25 19:52:07
result_TFHit %>%
as.data.frame() %>%
head(n = 10)
## TF_id mean_value TFPeak_number
## 1 LEC2 0.8809657 3223
## 2 GSE145387.VAL1.Col-0_seedling 0.5130745 9754
## 3 GSE145387.VAL2.Col-0_seedling 0.4457701 13039
## 4 GSE26722.REV.Col-0_seedling_10d-DEX-90min 0.3938903 8489
## 5 GSE48081.KAN1.Col-0_seedling_10d-rep1 0.4757618 13815
## 6 GSE94307.HEC1.Col-0_seedling_12d 0.4541106 9082
## 7 GSE142621.BRM.Col-0_seedling_BRIP1 0.3749682 8247
## 8 GSE122579.AT2G17950.Col-0_seedling 0.4619562 10908
## 9 GSE115358.AT2G14045.Col_seedling_TCP4-OE 0.4792326 13926
## 10 GSE80566.AT5G50360.Col-0_seedling_4d-DEX-6h 0.4728792 18597
## pvalue padj qvalue rank
## 1 0.000000e+00 0.000000e+00 0.000000e+00 1
## 2 3.218602e-181 2.233710e-178 2.188650e-179 2
## 3 1.398473e-130 9.691419e-128 6.339745e-129 3
## 4 7.509653e-128 5.196680e-125 2.553282e-126 4
## 5 2.645184e-122 1.827822e-119 7.194901e-121 5
## 6 1.168732e-121 8.064248e-119 2.649125e-120 6
## 7 4.881154e-120 3.363115e-117 9.483386e-119 7
## 8 1.848141e-113 1.271521e-110 3.141840e-112 8
## 9 4.868389e-110 3.344583e-107 7.356676e-109 9
## 10 7.733550e-100 5.305215e-97 1.051763e-98 10可以看到这回LEC2明显地排在了第一位。跟前面的findIT_TTPair一样,findIT_TFHit结果也包含着多列信息。第1列是TF_id,其按照mean_value进行排序,mean_value越高的,会排在越前面。第2列是mean_value,代表的是感兴趣基因集和背景基因集RP均值之差。第3列是TFPeak_number,代表的是数据库中该TF的binding site 数目。第4列是p-value,其来源于用wilcox.test函数对两个基因集的RP分布进行统计推断的结果。第5,6列是矫正后的p-value。第7列是rank值。
在使用findIT_TFHit的时候,有一些注意事项。首先就是findIT_TFHit的基本原理来源于calcRP_TFHit,这就意味着calcRP_TFHit中的一些参数也会应用到这里来,包括scan_dist,decay_dist等,大家需要根据自己的基因组进行调整。其次就是在构建TF_GR_database的时候,因为我们关注的TF在基因集合中的RP分布,而不是每一个binding site对于基因的RP贡献,所以我们需要添加一列TF_id来表征每一个TF的binding site集合。换而言之,在这个函数中,每一个peak的location(feature_id)并不是我们所关注的,TF对应的location集合(TF_id)才是。
尽管相比于TF-target pair数据库,有更多的物种含有TF ChIP-seq binding site (TFBS)数据库。但是大部分非模式物种依旧没有一个完整的TFBS数据库可以使用。这时候为了得到TFBS位点,可以考虑用对应物种ATAC-seq的motif或者footprint scan的结果来作为TFBS。现在常用的motif scan的软件有GimmeMotifs、HOMER、MEME、MOODS等。用户可以自己选择软件进行call motif,然后将产生的bed文件读取成GRanges对象,并添加一列TF_id来代表你的TF。操作的步骤与前面读取Remap2020数据库类似。
为了方便演示,我在这里使用的是motifmatchr这个R包来对ATAC-seq的peak进行motif scan。
# 写个函数来整理提取出JASPAR2020中的内容
getJasparMotifs <- function(species = "Arabidopsis thaliana",
collection = "CORE", ...) {
opts <- list()
opts["species"] <- species
opts["collection"] <- collection
opts <- c(opts, list(...))
out <- TFBSTools::getMatrixSet(JASPAR2020::JASPAR2020, opts)
if (!isTRUE(all.equal(TFBSTools::name(out), names(out))))
names(out) <- paste(names(out), TFBSTools::name(out), sep = "_")
return(out)
}
motifs_JASPAR2020 <- getJasparMotifs()
# call motif
motif_ix <- matchMotifs(motifs_JASPAR2020,
ATAC_peak_GR,
genome = BSgenome.Athaliana.TAIR.TAIR9,
out = "positions",
bg = "genome")
motif_GR <- unlist(motif_ix)
motif_GR$TF_id <- gsub("(MA[0-9]{4}\\.[0-9]_)", "", names(motif_GR))
motif_GR
## GRanges object with 2147996 ranges and 2 metadata columns:
## seqnames ranges strand | score TF_id
## <Rle> <IRanges> <Rle> | <numeric> <character>
## MA0121.1_ARR10 Chr1 1490-1497 - | 10.9848 ARR10
## MA0121.1_ARR10 Chr1 1904-1911 - | 10.9848 ARR10
## MA0121.1_ARR10 Chr1 33532-33539 - | 10.9848 ARR10
## MA0121.1_ARR10 Chr1 55227-55234 - | 11.1123 ARR10
## MA0121.1_ARR10 Chr1 62176-62183 - | 11.1123 ARR10
## ... ... ... ... . ... ...
## MA1329.2_ZHD1 Chr5 26876218-26876230 - | 18.9609 ZHD1
## MA1329.2_ZHD1 Chr5 26879928-26879940 - | 19.6284 ZHD1
## MA1329.2_ZHD1 Chr5 26880196-26880208 - | 19.6284 ZHD1
## MA1329.2_ZHD1 Chr5 26889355-26889367 - | 20.1565 ZHD1
## MA1329.2_ZHD1 Chr5 26901566-26901578 - | 19.1066 ZHD1
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsset.seed(19960203)
result_TFHit <- findIT_TFHit(input_genes = LEC2_target,
Txdb = Txdb,
TF_GR_database = motif_GR,
scan_dist = 2e4,
decay_dist = 1e3)
## >> preparing gene features information... 2021-09-25 19:52:53
## >> some scan range may cross Chr bound, trimming... 2021-09-25 19:52:53
## >> calculating p-value for each TF, which may be time consuming... 2021-09-25 19:52:54
## >> merging all info together... 2021-09-25 19:53:26
## >> done 2021-09-25 19:53:26
result_TFHit
## # A tibble: 465 x 7
## TF_id mean_value TFPeak_number pvalue padj qvalue rank
## <chr> <dbl> <dbl> <dbl> <dbl> <lgl> <dbl>
## 1 LEC2 0.142 2904 4.95e-63 2.30e-60 NA 1
## 2 FUS3 0.160 3383 5.45e-62 2.53e-59 NA 2
## 3 ABI3 0.161 2370 9.02e-58 4.18e-55 NA 3
## 4 DOF5.3 0.282 23535 1.76e-52 8.13e-50 NA 4
## 5 AT1G69570 0.369 31145 1.43e-49 6.61e-47 NA 5
## 6 AT5G66940 0.429 37990 1.42e-48 6.53e-46 NA 6
## 7 OBP3 0.559 51771 5.08e-48 2.33e-45 NA 7
## 8 IDD2 0.262 24356 2.41e-47 1.10e-44 NA 8
## 9 OBP1 0.225 18538 6.58e-46 3.01e-43 NA 9
## 10 AT2G28810 0.175 17649 7.56e-44 3.45e-41 NA 10
## # ... with 455 more rows可以看到, LEC2仍旧是排名前列的TF。跟它在一起的还有FUS3、ABI3等,这些都是跟ATAC样本,即体细胞胚发生,相关的TF。
在使用ATAC motif scan的结果作为findIT_TFHit输入的时候,有一些注意事项。首先就是上面用作示例的motifmatchr实际上是MOODS的R语言封装,这意思着motifmatchr的结果跟MOODS很类似,而MOODS虽然运行速度很快,但是也有文章报道说它会产生过多的TFBS结果。如果想要TFBS结果更加地准确,可以考虑用上面提到的GimmeMotifs。当然,大家也可以自己查阅文献,来选择自己认为合适的motif scan软件,也可以使用那些find Footprint的软件,只要你最终的结果能变成类似于上面一样的有TF_id列的GRange对象即可。
还有就是由于ATAC-seq的length不同,有些甚至有数千bp,这时候为了提高TFBS鉴定的准确性,可以考虑resize一下peak文件。像GimmeMoitf或者MEME在鉴定TFBS前似乎都是要求你resize下peak的。事实上不管是挑选软件还是resize peak,我们最终目的都是更加准确地鉴定有功能的TFBS、降低噪音。在R语言中的resize peak可以用以下的函数:
# 设定GRanges的seqinfo
seqinfo(ATAC_peak_GR) <- seqinfo(BSgenome.Athaliana.TAIR.TAIR9)
# 根据peak中心进行resize
ATAC_peak_GR_resize <- resize(ATAC_peak_GR, width = 500, fix = "center")
## Warning in valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE): GRanges object contains 2 out-of-bound ranges located on sequence Chr5.
## Note that ranges located on a sequence whose length is unknown (NA) or
## on a circular sequence are not considered out-of-bound (use
## seqlengths() and isCircular() to get the lengths and circularity flags
## of the underlying sequences). You can use trim() to trim these ranges.
## See ?`trim,GenomicRanges-method` for more information.
# 修剪掉越过边界的部分
ATAC_peak_GR_resize <- trim(ATAC_peak_GR_resize)
ATAC_peak_GR
## GRanges object with 22994 ranges and 2 metadata columns:
## seqnames ranges strand | feature_id score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] Chr1 30-875 * | ATAC_1 0
## [2] Chr1 1392-1953 * | ATAC_2 0
## [3] Chr1 2164-3625 * | ATAC_3 0
## [4] Chr1 8268-8966 * | ATAC_4 0
## [5] Chr1 13654-14698 * | ATAC_5 0
## ... ... ... ... . ... ...
## [22990] Chr5 26969184-26969915 * | ATAC_22990 0
## [22991] Chr5 26970542-26971504 * | ATAC_22991 0
## [22992] Chr5 26972304-26973186 * | ATAC_22992 0
## [22993] Chr5 26974280-26974825 * | ATAC_22993 0
## [22994] Chr5 26975157-26975438 * | ATAC_22994 0
## -------
## seqinfo: 7 sequences (2 circular) from TAIR9 genome
ATAC_peak_GR_resize
## GRanges object with 22994 ranges and 2 metadata columns:
## seqnames ranges strand | feature_id score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] Chr1 203-702 * | ATAC_1 0
## [2] Chr1 1423-1922 * | ATAC_2 0
## [3] Chr1 2645-3144 * | ATAC_3 0
## [4] Chr1 8367-8866 * | ATAC_4 0
## [5] Chr1 13926-14425 * | ATAC_5 0
## ... ... ... ... . ... ...
## [22990] Chr5 26969300-26969799 * | ATAC_22990 0
## [22991] Chr5 26970773-26971272 * | ATAC_22991 0
## [22992] Chr5 26972495-26972994 * | ATAC_22992 0
## [22993] Chr5 26974303-26974802 * | ATAC_22993 0
## [22994] Chr5 26975048-26975502 * | ATAC_22994 0
## -------
## seqinfo: 7 sequences (2 circular) from TAIR9 genome使用数据库中的ChIP-seq TFBS和用ATAC motif scan各有优劣之处。使用数据库中的位点优点是可以带给你更加准确的TFBS,但是由于数据库中的TF ChIP-seq组织状态各异,并不能真实地反映你所研究样本的特征。不过如果你数据库中的TF类型足够多,那么你可能也可以发现一些有意思的事情。比如说你用某种试剂处理前后的差异基因去寻找数据库中富集的TF,然后发现这些TF都是在肿瘤细胞中所做的,那么就可能说明你这个试剂可能跟癌症发生相关。
而使用你自己样本ATAC-seq的motif scan结果,尽管并不能真实地反映对应TF的binding site,因为你这个位置虽然有motif,但TF并不一定结合在这里。但用对应样本的ATAC-seq数据在一定程度上更加能反应你所研究样本的特征。所以如果你的物种既有数据库也有ATAC的话,建议两种都试试。
大家在看上面结果的时候可能会对qvalue为NA感到奇怪,根据Github的issuse来看,原因似乎是这个
Not enough p-values to estimate the proportion of truly null tests. You can the run the package as follows:
q=qvalue(p,pi0 = 1)
作者建议的pi0 = 1其实就是等于BH procedure,类似于padj中的BH矫正。所以在遇到qvalue都为NA的时候,直接用padj中的就可以了。
对于既没有TF ChIP-seq数据库,也没有ATAC-seq数据的物种,也可以考虑用保守性预测得到的TFBS或者说干脆就是全基因组motif scan得到的TFBS。只要我们记住输入的TF_GR_database应该尽可能地反映真实的TF结合位点就行。
对于植物而言,plantTFDB/plantRegMap和PLAZA这两个数据库中收录了比较全的植物TFBS位点,大家可以自己下载进行测试。我的建议是多测试几个数据库,然后看看哪些TF是出现在最前面的。
这里我用的是plantRegmap中拟南芥的genomewide TFBS location文件。可以看到LEC2(AT1G28300)仍旧排名前列。
mcols(plantTFDB_TFBS_GR) <- mcols(plantTFDB_TFBS_GR)[, "Name", drop = FALSE]
colnames(mcols(plantTFDB_TFBS_GR)) <- "TF_id"
set.seed(19960203)
result_TFHit <- findIT_TFHit(input_genes = LEC2_target,
Txdb = Txdb,
TF_GR_database = plantTFDB_TFBS_GR,
scan_dist = 2e4,
decay_dist = 1e3)
## >> preparing gene features information... 2021-09-25 19:53:26
## >> some scan range may cross Chr bound, trimming... 2021-09-25 19:53:27
## >> calculating p-value for each TF, which may be time consuming... 2021-09-25 19:53:28
## >> merging all info together... 2021-09-25 19:54:15
## >> done 2021-09-25 19:54:15
result_TFHit %>%
as.data.frame() %>%
head(n = 10)
## TF_id mean_value TFPeak_number pvalue padj qvalue
## 1 AT3G26790 0.17392689 6342 8.826010e-34 5.286780e-31 2.430547e-31
## 2 AT1G49480 0.53944698 108122 5.069598e-29 3.031619e-26 6.980446e-27
## 3 AT1G28300 0.11004602 4923 4.411664e-27 2.633764e-24 4.049682e-25
## 4 AT3G55370 0.17622342 46608 2.268201e-15 1.351848e-12 1.561569e-13
## 5 AT2G45660 0.74108874 159915 3.862282e-15 2.298058e-12 2.127226e-13
## 6 AT4G38000 0.20284547 56674 4.535187e-13 2.693901e-10 2.081535e-11
## 7 AT1G69570 0.14194568 35950 2.580157e-11 1.530033e-08 1.015051e-09
## 8 AT5G42520 0.87641468 81372 4.751911e-11 2.813131e-08 1.635754e-09
## 9 AT4G14410 0.05394649 5086 1.659576e-10 9.808094e-08 5.078019e-09
## 10 AT5G45300 0.05249233 5028 2.554286e-10 1.507029e-07 7.034110e-09
## rank
## 1 1
## 2 2
## 3 3
## 4 4
## 5 5
## 6 6
## 7 7
## 8 8
## 9 9
## 10 10Guandong Shang
PhD Candidate
My research interests include rstats, epigenomics, bioinformatics and evo-devo.