Intro_FindIT2:calcRP module
前两篇文章我分别介绍了mmAnno和peakGeneCormodule,这篇文章我来介绍FindIT2的第三个模块:calcRP。calcRP这个模块的思想脱胎于刘小乐老师的几篇文章1 2 3. 总体的思想就是大部分基因周围有着许多顺式调控元件,而这些调控元件上有着许多转录调节因子的结合位点。正是这些转录调节因子和顺序作用元件共同调控了基因的表达。如果我们将基因TSS周围元件的状态进行整合计算,然后对应得到一个调控潜能分数(regulatory potential,RP),我们就可以用这个RP来代表对应基因周围的染色质状态。
在我看来,将周围的染色质状态整合为一个分数有几个好处:首先最大的好处就是我们在处理染色质数据的时候我们不再面对的是peak,而是基因。不要小瞧这个优点,由于peak自身并没有注释信息,如果我们想要对聚类、差异分析等过程得到的功能性peak进行探究或者展示,我们就只能去将进行注释,用注释到的基因来作为其功能的表征。但在前两篇文章中我已经不断地强调了在某些时候最近基因注释原则的缺陷性,你大规模注释得到的对应基因不一定真的就是peak所调节的那个基因。而如果我们将基因周围的peak整合为一个RP分数,我们就可以对这个RP分数进行聚类,那么得到的结果就非常具有解释性,因为你直接得到的是XX基因的RP在几组样本中呈现的是XX趋势。
除了对得到的RP进行多样本聚类以外,如果你有非常多组织的染色质数据,你就可以用RP分数来鉴定细胞或者组织特异的marker基因4。根据我自己分析的结果来看,用RP来探究发现细胞或者组织特异的marker基因还是能够得到一些很有意思的结论的。同时,由于RP代表了基因周围染色质状态的一个概述,对应是一个基因层面的分数,其还可以作为一些机器学习方面的参数,尤其是涉及到基因表达层面的时候。更重要的是RP分数这个思想是后续的几个模块的基础,我会用RP来进行ChIP-seq和RNA-seq的联合分析,用RP来寻找重要的转录因子。
calcRP module总共有三个函数:calcRP_coverage是通过ATAC/H3K27ac的bw文件来计算每个基因RP的,calcRP_region则是通过mm_geneScan的ATAC/H3K27ac peak多对多注释以及对应peak的count文件来计算RP,而第三个函数calcRP_TFHit则是通过TF ChIP-seq的peak文件来计算RP的。具体的解释及应用我会在后面详细写出。
之所以强调是ATAC或者H3K27ac的样本,是因为除了一些异染色质的组蛋白修饰外,很多激活型的组蛋白修饰的genomic location也仍旧是局限在基因周围的,并不能标记到远端基因间区,也不能作为enhancer的标记。如果用这些激活型的组蛋白,比如说H3K36me3等来计算RP,是会损失很多信息的。
Pre-preparation
这次的测试数据仍旧来自于FindIT2包本身,包含了一个ATAC的bw文件,一个ATAC的多时间点的mergePeak文件及对应的count矩阵和一个TF的ChIP-seq peak文件。
首先我们需要加载对应的包以及数据。
# 加载包
library(FindIT2)
library(TxDb.Athaliana.BioMart.plantsmart28)
library(ggplot2)
Txdb <- TxDb.Athaliana.BioMart.plantsmart28
seqlevels(Txdb) <- paste0("Chr", c(1:5, "M", "C"))# 加载相关数据
bwFile <- system.file("extdata", "E50h_sampleChr5.bw", package = "FindIT2")
ATAC_peak_path <- system.file("extdata", "ATAC.bed.gz", package = "FindIT2")
ATAC_peak_GR <- loadPeakFile(ATAC_peak_path)
ChIP_peak_path <- system.file("extdata", "ChIP.bed.gz", package = "FindIT2")
ChIP_peak_GR <- loadPeakFile(ChIP_peak_path)
# 加载矩阵数据
data("ATAC_normCount")calcRP_coverage
大家可能会对前面说的
如果我们将基因TSS周围元件的状态进行整合计算,然后对应得到一个调控潜能分数(regulatory potential,RP),我们就可以用这个RP来代表对应基因周围的染色质状态`
感觉有点奇怪,所谓的整合到底是什么意思呢。事实上,这里的整合就是把你基因TSS附近的染色质分数给直接加和。比如你基因TSS附近的染色质分数有1,2,3,6,那么基因TSS的总和就是12。但是我们怎么定义染色质分数呢,在calcRP_coverage中,染色质分数就是TSS临近每个bp上的score值。比如我们定义TSS上下10bp为计算RP的范围,那么最终的RP就是这20bp score的和。因为calcRP_coverage的输入文件是bigwig类型,你可以认为这个score就是每个bp上的ATAC count值,如果你在bigwig生成过程中用了什么BPM、CPM等矫正操作,那么这个score就是矫正以后的count值了。
但事实上,这里的加和并不是简单的直接加和,而是一个权重的加和,即每个bp对于最终的RP score是有不同的贡献的。已经有很多的文献认为权重,即贡献大小是会随着距离衰减的,并不是始终一样的。这也非常符合我们的生物学常识,距离远的位置对于你基因的调控自然没有距离近的位置对于你基因的调控大。但衰减的程度又是怎么样的呢,在刘小乐老师的文章中,他们对于衰减函数的定义也经过了多番修改,但始终不变的是这个函数一定是一个指数衰减的函数。在calcRP_coverage中,我使用的是他们之前的一个函数,即
\[ RP_{gene_{g}}=\sum_{i\in[t_g-L,tg+L]}w_iS_i \]
\[
w_i=\frac {2e^{-\mu d}} {1+e^{-\mu d}}
\]
公式中的 \(t_g\) 是指 \(gene_g\) 的TSS位置,\(L\)是指你设置的上下游计算范围,对应的 \(i\) 就是在这个计算范围内的每个bp。\(w_i\) 就是每个bp对应的权重,\(S_i\)就是每个bp上的score。最终 \(gene_g\) 的RP就是每个bp的score乘上对应权重的和。举个例子,我们设定我们的计算范围为上下游1bp,每个bp的score分别为2,1,1,对应的权重为0.5,1,0.5。那么最终的RP就是 2*0.5 + 1*1 + 1*0.5 = 1 + 1 + 0.5 = 2.5。
我们前面提到权重函数是一个指数衰减函数,第二个公式就是所谓的权重函数。\(e\)是一个数学常数,而\(d\)是第\(i\)个bp到TSS的绝对距离与\(L\)相除的结果,即\(d = |i − t_{g}|/L\)。\(\mu\)控制的是权重的衰减速率,定义为\(\mu = -ln\frac{1}{3} * (L/\Delta)\)。这里的 \(\Delta\) 值是半衰期距离,即距离TSS \(\Delta\) 的那个bp,其权重刚好为 \(\frac{1}{2}\) 。
这个公式可能看的有点云里雾里,让我们来实际举个例子,假设我们设置上下游计算范围为 100kb,然后我们设置半衰期距离为10kb,那么我们设置不同的距离
(mu = -log(1/3) * (1e5/1e4))
## [1] 10.98612
# 分别距离TSS -100,-50,0,10kb,即-1e5,-5e4,0,1e4
w_vector <- vector(mode = "numeric", 4)
i_vector <- c(-1e5, -5e4, 0, 1e4)
for (index in seq_len(4)){
d = abs(i_vector[index]) / 1e5
w_vector[index] = (2 * exp(- mu * d)) / (1 + exp(- mu * d))
}
w_vector
## [1] 0.0000338696 0.0081967213 1.0000000000 0.5000000000可以看到,在距离为1e4,即10kb的时候,刚好权重为1/2,而在距离的0的时候,权重为1,在距离为1e5的时候,权重几乎为0了。如果这个还不是很直观的话,我们可以用图像来表示。从图中中我们可以直观地看出,1e5和5e4的权重几乎是一样的,即在距离TSS比较远的时候权重的变化速率是比较慢的,而越靠近TSS,权重的增加速率就会非常地快了。
ggplot(data.frame(x = c(-1e5, 1e5)), aes(x = x)) +
stat_function(fun = function(x) (2 * exp(- mu * abs(x) / 1e5)) / (1 + exp(- mu * abs(x) / 1e5))) +
theme_bw() +
geom_vline(xintercept = i_vector)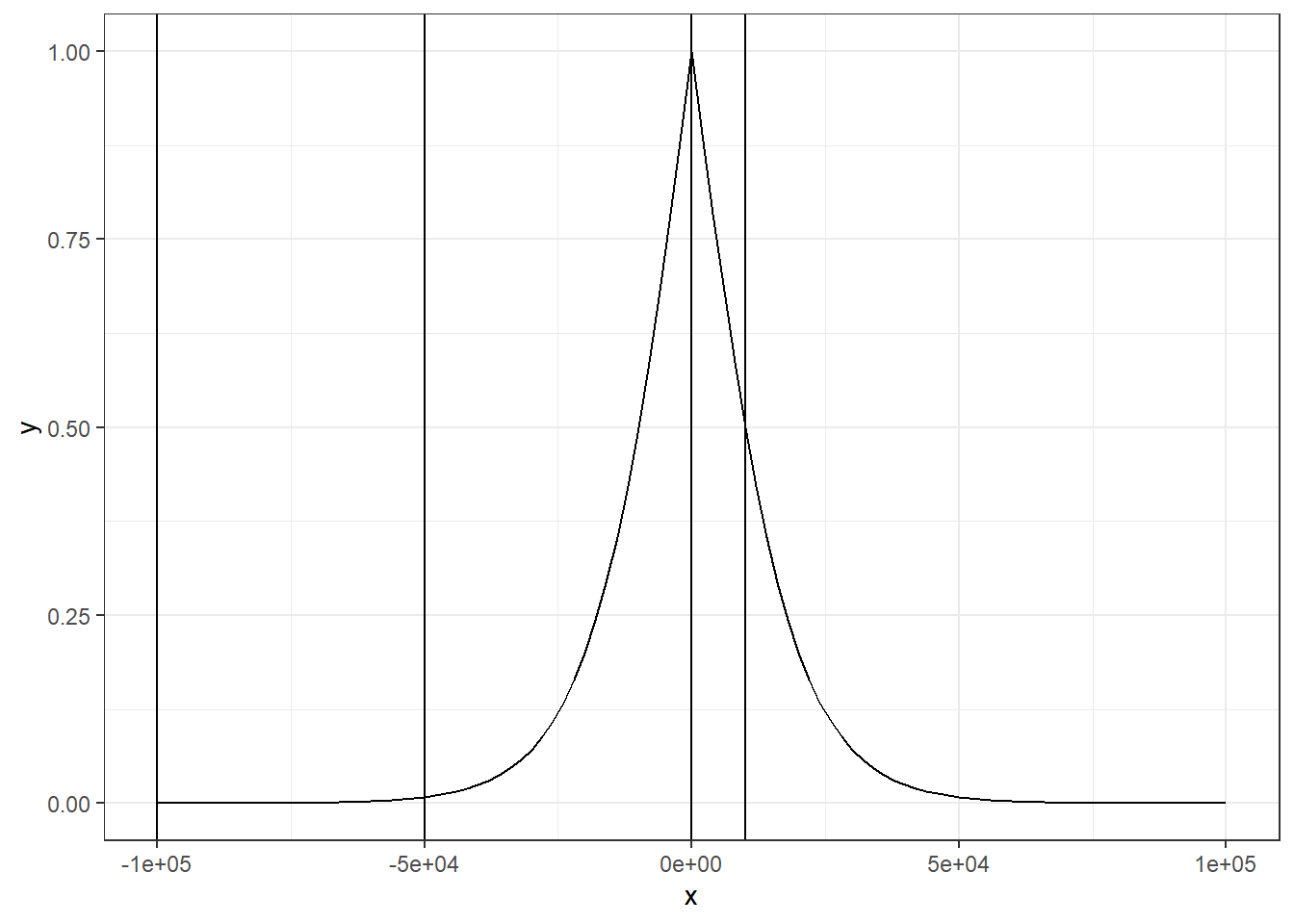
在讲完了基本概念之后,我们就可以直接对测试数据进行RP计算了。calcRP_coverage支持的输入文件类型是bigwig格式。bigwig是大部分文章数据在投递到GEO数据库的时候会选择的格式之一,通过calcRP_coverage,用户可以快速方便地对文章里面的样本计算基因的RP。我这里用的是包附带的bigwig文件,只用了拟南芥上第5条染色体的数据。可以看到产生的结果是一个两列的数据框,一列是gene_id,一列是基因对应的RP。如果这个基因的sumRP越高,越说明这个基因附近的染色质越开放。
# bwFile只支持单个文件的路径
RP_df <- calcRP_coverage(
bwFile = bwFile,
Txdb = Txdb,
decay_dist = 1e3,
scan_dist = 2e4,
Chrs_included = "Chr5"
)
## >> preparing gene features information... 2021-08-25 17:05:56
## >> some scan range may cross Chr bound, trimming... 2021-08-25 17:05:57
## >> preparing weight info... 2021-08-25 17:05:57
## >> loadingE50h_sampleChr5.bwinfo... 2021-08-25 17:05:57
## ------------
## >> extracting and calcluatingChr5signal... 2021-08-25 17:05:57
## >> dealing withChr5left gene signal... 2021-08-25 17:05:58
## >> normingChr5RP accoring to the whole Chr RP... 2021-08-25 17:05:59
## >> merging all Chr RP together... 2021-08-25 17:05:59
## >> done 2021-08-25 17:05:59
head(RP_df)
## gene_id sumRP
## 1 AT5G01050 -0.8437482
## 2 AT5G01060 0.6576913
## 3 AT5G01070 3.5641593
## 4 AT5G01075 3.2401437
## 5 AT5G01080 1.6852928
## 6 AT5G01090 -1.3254554大家可能会对这个函数的一些参数选择有一些疑问,在这里我再详细地展开讲下。首先就是bw的选择问题,大家可能会疑惑我应该用什么方式去产生bw呢,是使用deeptools呢还是UCSC呢,如果使用deeptools,我该使用什么样的矫正方法呢。实际上,如果你只是针对一个样本计算其RP的话,那么不管用什么都是一样的。但如果你有多组bw样本,想要计算每个基因在每个样本中的RP的话,你的bw文件应该是经过矫正的,且每个样本使用的矫正方法应该是一样的,而对于矫正方法的选择并没有太大关系。
除了常规使用的deeptools的bamCoverage以外,还可以考虑用MACS2自带的pileup函数或者在callpeak的时候加入 -B –SPMR 参数。这样就会生成一个bedGraph文件,这个文件是一个
pileup signal file of 'fragment pileup per million reads',也算是矫正过的。然后你再用UCSC的bedGraphToBigWig就可以转变成bigwig了。根据刘小乐老师他们处理CistromeDB的源代码来看,似乎用的是后者方法。
除了bw文件以外,可能大家感到困惑的还有decay_dist和scan_dist了。scan_dist指的就是前面公式里面的 \(L\),即你选择的计算范围。在刘小乐老师的几篇文章中,他们对于小鼠和人类都设置的是100kb,即1e5,这个数值的来源似乎是TAD的范围。我这里拟南芥的数字设置的是2e4,因为在拟南芥Hi-C数据中5, 20kb是最长的作用范围了。不过这个数值对于最后的结果影响并不是很大,真正影响比较大的是decay_dist,即半衰期距离,即前面公式的\(\Delta\)。
这个参数控制了距离多少bp的权重达到了\(\frac{1}{2}\)。刘小乐老师的文章中是10kb,这个数值似乎来自于Hi-C的参数拟合,即在10kb左右,Hi-C的作用频率是达到了一半的。我这里拟南芥设置的是1e3,我个人觉得这个比较合理,因为作为一个小基因组物种,其作用的范围应该比较短,同时由于大部分拟南芥的ChIP-seq或者ATAC-seq的距离分布图,在1kb左右有一个次峰,所以我觉得1e3可能是一个比较合理的数值了。对于其他用户而言,可以根据自己的基因组大小适当调整范围。
还有就是Chrs_included和gene_included这两个参数了,如果你只想要计算单独几个基因的RP,那么就可以考虑在gene_included中选择对应的基因。如果你只想要计算某条染色体上的RP,就可以在Chrs_included中选择对应的基因。这两个参数对于结果影响不大,只是可以方便提升计算速度,因为如果你只选择了几条染色体,那么calcRP_coverage就不会去计算其他的了。
大家可能还会对前面RP_df里面sumRP的结果有点疑惑,明明前面的公式里面是加和,为什么会出现负值呢。因为`calcRP_coverage在计算RP之后,还对每条染色体上基因RP做了单独的矫正。因为每条染色体上的RP分布是不一样的,因为我们有时候想要看的是所有基因的RP分布,所以我们就需要根据每条染色体对RP进行矫正。矫正的过程就是把RP数值加1再取log得到log之后的值,然后把整条染色体上的logRP数值求平均,每个基因的矫正RP值就是logRP减去平均值。
\[ RP_{gene_g}' = log(RP_{gene_g} + 1) - \frac{1}{g}\sum_{1}^{g}(log(RP_{gene_g} + 1)) \] 这样矫正还有一个好处就是我们看基因的RP数值就可以立刻反应过来这个基因RP是高还是低,如果基因周围几乎没有染色质开放,那么其RP就会很低,反映到数值上来就是负值了。
大家可能还记得我前面提到RP的一个好处就是可以鉴定细胞或者组织特异的marker基因,这里我给出一个简单的操作。如果用户有很多的ATAC样本,分别根据每个样本的bigwig样本来得到RP,然后整合下,就得到了一个行为基因列为RP的矩阵。然后你可以每个样本中的每个基因减去其对应的行平均值/中位数,然后画个热图,看下其中数值最高的基因,就有可能是组织特异的了。
set.seed(20160806)
mt <- matrix(data = rnorm(20, mean = 2, sd = 3),
nrow = 5,
ncol = 4)
rownames(mt) <- paste0("gene", 1:5)
mt
## [,1] [,2] [,3] [,4]
## gene1 1.959472 4.1827209 0.1185374 3.0904326
## gene2 1.827178 3.0307240 2.6158794 3.3741778
## gene3 3.056564 5.6712762 -2.9783050 -3.1827982
## gene4 4.811699 3.9170313 4.1217227 0.1479771
## gene5 3.177273 -0.9004561 4.7939500 6.5865681
# 求每行的平均值,即基因在每个样本中的RP平均值
(RPSample_mean <- apply(mt, 1, mean))
## gene1 gene2 gene3 gene4 gene5
## 2.3377908 2.7119898 0.6416843 3.2496075 3.4143338
sweep(mt, 1, RPSample_mean, FUN = "-")
## [,1] [,2] [,3] [,4]
## gene1 -0.3783185 1.8449301 -2.21925339 0.7526418
## gene2 -0.8848117 0.3187342 -0.09611046 0.6621880
## gene3 2.4148800 5.0295919 -3.61998935 -3.8244825
## gene4 1.5620913 0.6674238 0.87211519 -3.1016303
## gene5 -0.2370605 -4.3147899 1.37961614 3.1722343calcRP_region
calcRP_region的基本原理跟前面calcRP_coverage差不多,也是计算基因TSS附近的染色质分数,然后进行加和,同时也考虑到了权重的问题。不过输入文件和权重公式有稍微的不同。首先是权重公式,这里的权重公式更加地简化易懂了。
\[ RP_{gene_{g}}=\sum_{p=1}^{k}RP_{peak_p, gene_g} \]
\[ RP_{peak_p, gene_g} = score_{peak_p} * 2^{\frac{-d_{i}}{d_0}} \]
每个基因的RP是由基因TSS附近的peak综合而来,然后peak的score就代表了peak的count数值,权重变成了一个很简单的\(2^{\frac{-d_{i}}{d_0}}\)函数,\(d_i\)就是第i个peak到基因TSS的距离,d0就是半衰期距离。我们可以再次用图来展示下这个函数。
ggplot(data.frame(x = c(-1e5, 1e5)), aes(x = x)) +
stat_function(fun = function(x) 2 ^ (-abs(x)/1e4)) +
theme_bw() +
geom_vline(xintercept = i_vector)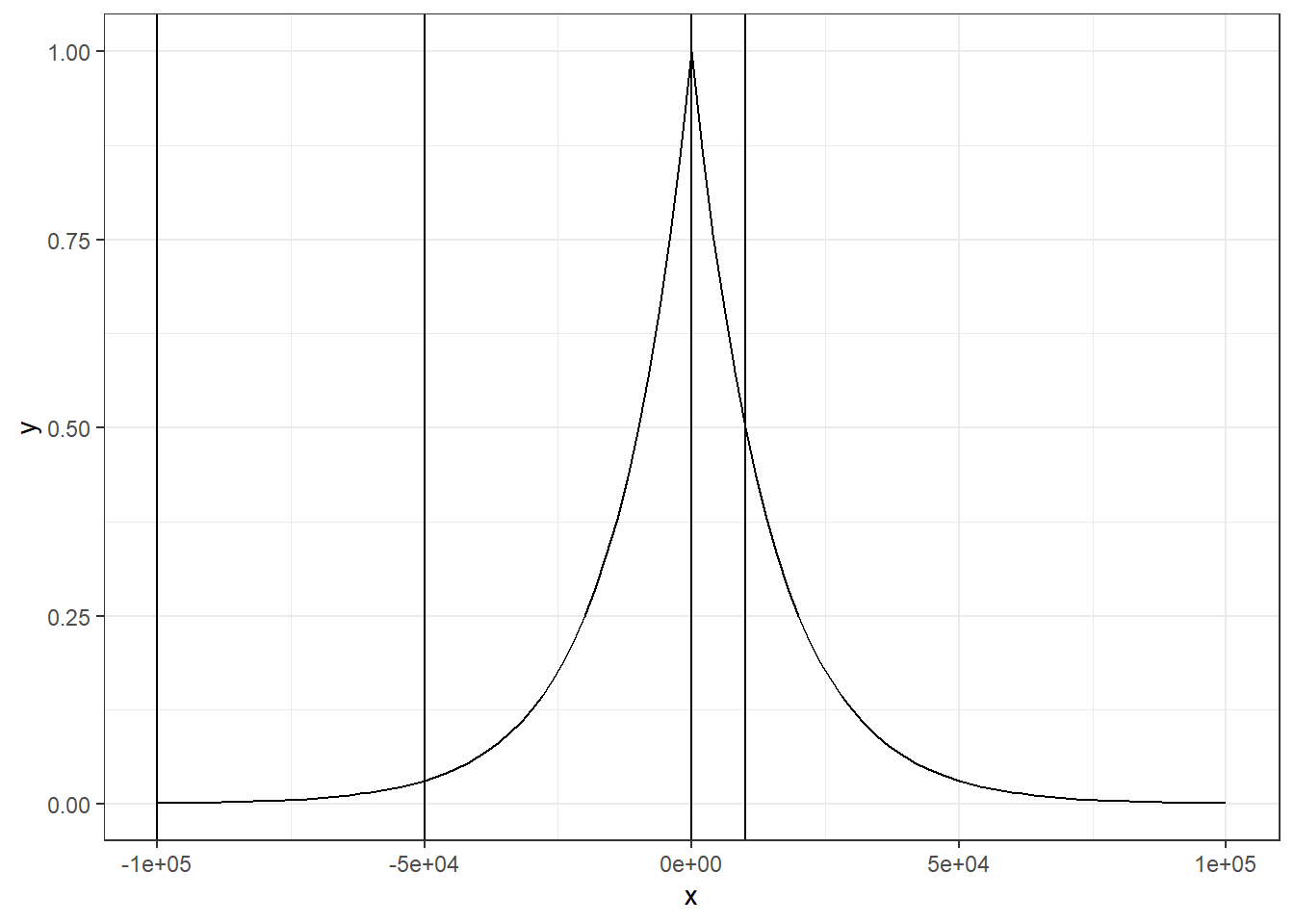
因为我们这里计算的单位不再是bp,而是peak了,所以我们就需要有对应的peak注释结果和count矩阵了。还记得我们之前mmAnno module里面的mm_geneScan么,我们这里的输入文件之一就是mm_geneScan的结果。
# 先计算得到peak注释文件
# mm_geneScan的upstream和downstream代替了前面calcRP_coverage的scan_dist
mmAnno_regionRP <- mm_geneScan(ATAC_peak_GR,
Txdb,
upstream = 2e4,
downstream = 2e4)
## >> checking seqlevels match... 2021-08-25 17:05:59
## >> your peak_GR seqlevel:Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## ------------
## annotatePeak using geneScan mode begins
## >> preparing gene features information and scan region... 2021-08-25 17:05:59
## >> some scan range may cross Chr bound, trimming... 2021-08-25 17:05:59
## >> finding overlap peak in gene scan region... 2021-08-25 17:05:59
## >> dealing with left peak not your gene scan region... 2021-08-25 17:05:59
## >> merging two set peaks... 2021-08-25 17:06:00
## >> calculating distance and dealing with gene strand... 2021-08-25 17:06:00
## >> merging all info together ... 2021-08-25 17:06:00
## >> done 2021-08-25 17:06:00
# 然后再根据注释文件和peak count矩阵来计算RP
calcRP_region(mmAnno = mmAnno_regionRP,
peakScoreMt = ATAC_normCount,
decay_dist = 1e3,
Txdb = Txdb,
Chrs_included = "Chr5",
log_transform = FALSE) -> regionRP
## >> calculating peakCenter to TSS using peak-gene pair... 2021-08-25 17:06:00
## >> pre-filling 446 noAssoc peak gene's RP with 0... 2021-08-25 17:06:01
## >> calculating RP using centerToTSS and peak score2021-08-25 17:06:01
## >> merging all info together 2021-08-25 17:06:03
## >> done 2021-08-25 17:06:03
regionRP
## A MultiAssayExperiment object of 2 listed
## experiments with user-defined names and respective classes.
## Containing an ExperimentList class object of length 2:
## [1] fullRP: RangedSummarizedExperiment with 69296 rows and 16 columns
## [2] sumRP: matrix with 7507 rows and 16 columns
## Functionality:
## experiments() - obtain the ExperimentList instance
## colData() - the primary/phenotype DataFrame
## sampleMap() - the sample coordination DataFrame
## `$`, `[`, `[[` - extract colData columns, subset, or experiment
## *Format() - convert into a long or wide DataFrame
## assays() - convert ExperimentList to a SimpleList of matrices
## exportClass() - save all data to files这里calcRP_region的输出结果不再是一个数据框了,而是一个MultiAssayExperiment对象。具体的MultiAssayExperiment对象的介绍可以参考Bioconductor上的手册。简单来说,MultiAssayExperiment是一个更大的容器,可以容纳Bioconductor的很多对象类型,包括像GRanges,ExpressionSet,SummarizedExperiment等。
如果你想要提取出像之前一样的RP数据框,你可以用
sumRP <- MultiAssayExperiment::assays(regionRP)$sumRP
head(sumRP)
## E5_0h_R1 E5_0h_R2 E5_4h_R1 E5_4h_R2 E5_8h_R1 E5_8h_R2 E5_16h_R1
## AT5G01010 222.2079 212.7673 218.1653 213.6052 249.9372 206.0400 254.9596
## AT5G01015 208.7814 203.1862 203.0282 203.9045 224.0673 192.7383 240.3690
## AT5G01020 433.2978 439.1758 414.0660 446.9142 370.8003 419.2071 507.6845
## AT5G01030 417.1240 422.5180 395.8684 429.5824 349.7193 401.1142 486.7315
## AT5G01040 398.0912 398.0977 321.6067 351.3717 243.5476 236.7626 367.2205
## AT5G01050 269.0832 285.3329 117.0695 102.0756 109.3351 101.0077 135.9758
## E5_16h_R2 E5_24h_R1 E5_24h_R2 E5_48h_R1 E5_48h_R2 E5_48h_R3 E5_72h_R1
## AT5G01010 294.6202 271.1531 262.5603 216.3946 214.0887 226.4749 183.1970
## AT5G01015 270.5445 248.4688 241.9568 196.3324 194.7707 203.5344 168.8261
## AT5G01020 524.0937 489.1091 473.2037 338.7931 370.7956 356.9675 332.7021
## AT5G01030 499.3811 467.3236 452.1052 322.8229 353.5329 339.1584 320.1101
## AT5G01040 342.5372 360.6502 355.6829 293.5963 295.9778 290.2336 379.6624
## AT5G01050 139.9906 154.1518 129.9962 134.2388 113.4725 138.8942 161.2767
## E5_72h_R2 E5_72h_R3
## AT5G01010 201.5551 232.2543
## AT5G01015 188.3035 213.0661
## AT5G01020 382.2974 405.6337
## AT5G01030 366.3332 389.1395
## AT5G01040 342.0031 381.4891
## AT5G01050 174.7018 176.5979如果你想要知道具体的每个peak对于每个基因在每个样本中的RP贡献,你可以使用
fullRP <- MultiAssayExperiment::assays(regionRP)$fullRP
head(fullRP)
## E5_0h_R1 E5_0h_R2 E5_4h_R1 E5_4h_R2
## WFX_peak_19629:AT5G01010 3.2505507 2.94366371 4.91215176 3.2210557
## WFX_peak_19630:AT5G01010 28.3673260 25.49941664 33.09901392 26.4569161
## WFX_peak_19631:AT5G01010 11.3199591 8.66507551 7.69205730 10.1043687
## WFX_peak_19632:AT5G01010 140.0066186 135.72131106 135.00039379 133.1331029
## WFX_peak_19633:AT5G01010 38.7559482 39.48821374 37.10950497 40.2704327
## WFX_peak_19634:AT5G01010 0.1620359 0.09696071 0.05939023 0.1040209
## E5_8h_R1 E5_8h_R2 E5_16h_R1 E5_16h_R2
## WFX_peak_19629:AT5G01010 4.63291194 3.40978941 3.4267032 5.0733396
## WFX_peak_19630:AT5G01010 33.35699057 35.98214354 31.2959400 37.9690563
## WFX_peak_19631:AT5G01010 10.00585368 10.17992743 15.1914895 20.1385141
## WFX_peak_19632:AT5G01010 169.10492510 118.32576124 158.9697269 184.2512264
## WFX_peak_19633:AT5G01010 32.53901650 37.84312560 45.6184227 46.7559771
## WFX_peak_19634:AT5G01010 0.08068857 0.09220271 0.1290225 0.1258721
## E5_24h_R1 E5_24h_R2 E5_48h_R1 E5_48h_R2
## WFX_peak_19629:AT5G01010 4.7636561 3.6203395 3.1645878 3.61212739
## WFX_peak_19630:AT5G01010 41.7795900 37.5422424 27.0006566 32.58453567
## WFX_peak_19631:AT5G01010 16.1608755 14.0341872 8.6911928 13.21967538
## WFX_peak_19632:AT5G01010 164.3433125 164.7184992 147.3949774 131.34350008
## WFX_peak_19633:AT5G01010 43.6546465 42.2005830 29.7367518 32.99092356
## WFX_peak_19634:AT5G01010 0.1348949 0.1294783 0.1536341 0.07837431
## E5_48h_R3 E5_72h_R1 E5_72h_R2 E5_72h_R3
## WFX_peak_19629:AT5G01010 3.44344427 2.7374503 2.61457722 2.6626243
## WFX_peak_19630:AT5G01010 28.19522111 25.5527166 27.32943247 32.1836089
## WFX_peak_19631:AT5G01010 17.57139155 11.1322592 9.29903999 13.0251325
## WFX_peak_19632:AT5G01010 145.44860373 113.7699899 127.73896923 147.9226052
## WFX_peak_19633:AT5G01010 31.46275054 29.5742561 34.19603669 35.9567241
## WFX_peak_19634:AT5G01010 0.09615333 0.0841232 0.06690242 0.1683909这里的decay_dist和Chrs_included和前面calcRP_coverage中的参数意义一样。但calcRP_region也有一些需要额外注意的地方,首先就是log_transform参数。默认情况下,log_transform是FALSE,RP并不会被log,也不会针对染色体而矫正。如果你想要达到之前calcRP_coverage的效果,可以考虑log_transform设置为TRUE。在设置log_transform为TRUE的时候,如果想要计算组织特异的基因,可以考虑RP减样本平均值/中位数,如果log_transform为FALSE,可以考虑RP除平均值/中位数。还有就是peakCount矩阵的矫正问题,如果你的peakcount矩阵来源于多个样本,请一定要矫正你的count值,你可以用简单的CPM、FPKM,也可以用DESeq2或者edgeR来矫正原始的count,在这里我不做推荐。
calcRP_TFHit
calcRP_TFHit严格来说并不是用来计算基因周围的染色质状态的,其是用来计算基因周围TF的Hit情况的。针对一个TF的ChIP-seq数据,如果一个基因周围有越多的TF binding site,那么其就越有可能是这个TF的靶基因。换句话说,其是用来作为TF target的筛选的。但是其背后的理论依据是与前面两个函数一致的,公式也是用的一样的。
\[ RP_{gene_{g}}=\sum_{p=1}^{k}RP_{peak_p, gene_g} \]
\[
RP_{peak_p, gene_g} = score_{peak_p} * 2^{\frac{-d_{i}}{d_0}}
\]
只不过需要注意的是,在calcRP_TFHit使用这个公式的时候,函数一般会认为peak score大家都为1,即每个peak对于最终RP的贡献只取决于这个peak到基因TSS的距离。这么做的理由似乎是因为TF ChIP-seq的peak对于基因的作用强度本来就是距离越近越强的,所以加不加score都是一样的。不过如果用户想要考虑自己对于peak的置信度,也可以自己手动在peak_GR中添加一列feature_score,这样的话,calcRP_TFHit就会把原来的1替换成用户对应的score了。
不管是刘小乐老师或者之前的一些文章,作者都是将score变成1的,即peak对于最终RP的贡献只取决于这个peak到基因TSS的距离。描述的理由也的确是我提到的这个原因,即加入了score之后,结果并不会有太大的区别。但我个人觉得上面的原因是生物体的内源性结果,在TF ChIP-seq的建库过程中,还受到了很多因素的影响的,比如说抗体、Binding site本身的genomic sequence组成等等。更重要的是,很多时候TF ChIP-seq的实验用的并不是内源抗体,而是带了Flag标签的,又或者说TF ChIP-seq是在GR等诱导剂下完成的,这些可能都使得上面那个假设并不成立。所以我在
calcRP_TFHit函数中还提供了feature_score这个选项让大家可以将自己的置信度考虑在内。但需要注意的一点是,feature_score的数量级最好不要差距太大,不然最后的RP结果会失真。
# 你可以手动添加一列feature_score
# 或者把score这里给替换成feature_score,只要你认为这一列的确代表了你对于peak的置信程度
ChIP_peak_GR
## GRanges object with 4288 ranges and 2 metadata columns:
## seqnames ranges strand | feature_id score
## <Rle> <IRanges> <Rle> | <character> <numeric>
## [1] Chr5 6236-6508 * | peak_14125 27
## [2] Chr5 7627-8237 * | peak_14126 51
## [3] Chr5 9730-10211 * | peak_14127 32
## [4] Chr5 12693-12867 * | peak_14128 22
## [5] Chr5 13168-14770 * | peak_14129 519
## ... ... ... ... . ... ...
## [4284] Chr5 26937822-26938526 * | peak_18408 445
## [4285] Chr5 26939152-26939267 * | peak_18409 21
## [4286] Chr5 26949581-26950335 * | peak_18410 263
## [4287] Chr5 26952230-26952558 * | peak_18411 30
## [4288] Chr5 26968877-26969091 * | peak_18412 26
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
# colnames(mcols(ChIP_peak_GR))[2] <- "feature_score"
mmAnno_TFHit <- mm_geneScan(ChIP_peak_GR,
Txdb,
upstream = 2e4,
downstream = 2e4)
## >> checking seqlevels match... 2021-08-25 17:06:04
## >> your peak_GR seqlevel:Chr5...
## >> your Txdb seqlevel:Chr1 Chr2 Chr3 Chr4 Chr5 ChrM ChrC...
## Good, your Chrs in peak_GR is all in Txdb
## ------------
## annotatePeak using geneScan mode begins
## >> preparing gene features information and scan region... 2021-08-25 17:06:04
## >> some scan range may cross Chr bound, trimming... 2021-08-25 17:06:04
## >> finding overlap peak in gene scan region... 2021-08-25 17:06:04
## >> dealing with left peak not your gene scan region... 2021-08-25 17:06:04
## >> merging two set peaks... 2021-08-25 17:06:04
## >> calculating distance and dealing with gene strand... 2021-08-25 17:06:04
## >> merging all info together ... 2021-08-25 17:06:04
## >> done 2021-08-25 17:06:04如果你设置了report_fullInfo为TRUE,那么产生的结果就是一个GRanges对象,其代表了每个peak对于对应基因的RP贡献。
fullRP_hit <- calcRP_TFHit(mmAnno = mmAnno_TFHit,
Txdb = Txdb,
decay_dist = 1000,
report_fullInfo = TRUE)
## >> calculating peakCenter to TSS using peak-gene pair... 2021-08-25 17:06:04
## >> calculating RP using centerToTSS and TF hit 2021-08-25 17:06:05
## >> merging all info together 2021-08-25 17:06:05
## >> done 2021-08-25 17:06:05
fullRP_hit
## GRanges object with 50662 ranges and 6 metadata columns:
## seqnames ranges strand | feature_id score gene_id
## <Rle> <IRanges> <Rle> | <character> <numeric> <character>
## [1] Chr5 6236-6508 * | peak_14125 27 AT5G01010
## [2] Chr5 7627-8237 * | peak_14126 51 AT5G01010
## [3] Chr5 9730-10211 * | peak_14127 32 AT5G01010
## [4] Chr5 12693-12867 * | peak_14128 22 AT5G01010
## [5] Chr5 13168-14770 * | peak_14129 519 AT5G01010
## ... ... ... ... . ... ... ...
## [50658] Chr5 26949581-26950335 * | peak_18410 263 AT5G67630
## [50659] Chr5 26952230-26952558 * | peak_18411 30 AT5G67630
## [50660] Chr5 26968877-26969091 * | peak_18412 26 AT5G67630
## [50661] Chr5 26952230-26952558 * | peak_18411 30 AT5G67640
## [50662] Chr5 26968877-26969091 * | peak_18412 26 AT5G67640
## distanceToTSS centerToTSS RP
## <numeric> <integer> <numeric>
## [1] -1174 1310 0.40332088
## [2] -2565 2870 0.13678671
## [3] -4668 4908 0.03330771
## [4] -7631 7718 0.00474953
## [5] -8106 8907 0.00208318
## ... ... ... ...
## [50658] 19058 19435 1.41085e-06
## [50659] 16835 16999 7.63468e-06
## [50660] 302 409 7.53145e-01
## [50661] 18082 18246 3.21667e-06
## [50662] 1549 1656 3.17318e-01
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths如果你想要每个基因的RP综合结果,可以选择
metadata(fullRP_hit)$peakRP_gene
## # A tibble: 6,783 x 4
## gene_id withPeakN sumRP RP_rank
## <chr> <int> <dbl> <dbl>
## 1 AT5G67190 14 4.31 1
## 2 AT5G05140 25 3.56 2
## 3 AT5G62280 12 3.51 3
## 4 AT5G44380 10 3.38 4
## 5 AT5G58750 13 3.36 5
## 6 AT5G66590 13 3.36 6
## 7 AT5G02760 12 3.24 7
## 8 AT5G41071 8 3.23 8
## 9 AT5G64100 18 3.21 9
## 10 AT5G24030 12 3.19 10
## # ... with 6,773 more rows或者设置report_fullInfo为FALSE。
calcRP_TFHit(mmAnno = mmAnno_TFHit,
Txdb = Txdb,
decay_dist = 1000,
report_fullInfo = FALSE)
## >> calculating peakCenter to TSS using peak-gene pair... 2021-08-25 17:06:05
## >> calculating RP using centerToTSS and TF hit 2021-08-25 17:06:06
## >> merging all info together 2021-08-25 17:06:06
## >> done 2021-08-25 17:06:06
## # A tibble: 6,783 x 4
## gene_id withPeakN sumRP RP_rank
## <chr> <int> <dbl> <dbl>
## 1 AT5G67190 14 4.31 1
## 2 AT5G05140 25 3.56 2
## 3 AT5G62280 12 3.51 3
## 4 AT5G44380 10 3.38 4
## 5 AT5G58750 13 3.36 5
## 6 AT5G66590 13 3.36 6
## 7 AT5G02760 12 3.24 7
## 8 AT5G41071 8 3.23 8
## 9 AT5G64100 18 3.21 9
## 10 AT5G24030 12 3.19 10
## # ... with 6,773 more rows大家可以看到,RP的综合结果是一个排序的结果,描述了这个基因周围的peak数目,基因最后的RP,以及它的排名,排名越高,越说明这个基因有可能是TF的靶基因。我们也可以把fullRP_hit作为integrate_ChIP_RNA的输入,从而做ChIP-seq和RNA-seq的整合,来提高TF靶基因预测的准确度。这部分留待下一篇再说。
Reference
Wang, S., Zang, C., Xiao, T., Fan, J., Mei, S., Qin, Q., Wu, Q., Li, X., Xu, K., He, H.H., et al. (2016). Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 26, 1417–1429.↩︎
Tang, Q., Chen, Y., Meyer, C., Geistlinger, T., Lupien, M., Wang, Q., Liu, T., Zhang, Y., Brown, M., and Liu, X.S. (2011). A Comprehensive View of Nuclear Receptor Cancer Cistromes. Cancer Res 71, 6940–6947.↩︎
Wang, S., Sun, H., Ma, J., Zang, C., Wang, C., Wang, J., Tang, Q., Meyer, C.A., Zhang, Y., and Liu, X.S. (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8, 2502–2515.↩︎
Wang, S., Zang, C., Xiao, T., Fan, J., Mei, S., Qin, Q., Wu, Q., Li, X., Xu, K., He, H.H., et al. (2016). Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 26, 1417–1429.↩︎
Liu, C., Wang, C., Wang, G., Becker, C., Zaidem, M., and Weigel, D. (2016). Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 26, 1057–1068.↩︎
Guandong Shang
PhD Candidate
My research interests include rstats, epigenomics, bioinformatics and evo-devo.